LA FORMALIZACIÓN DEL CONSENTIMIENTO INFORMADO EN INVESTIGACIÓN Y LA PÉRDIDA SENTIMENTAL DEL PACIENTE
FORMALIZATION OF INFORMED CONSENT IN RESEARCH AND THE PATIENT'S EMOTIONAL LOSS
A FORMALIZAÇÃO DO CONSENTIMENTO INFORMADO EM PESQUISA E A PERDA SENTIMENTAL DO PACIENTE
Antonio Pardo-Caballos1, Luis Enrique Echarte-Alonso2
1 Universidad de Navarra, España.
apardo@unav.es
2 Universidad de Navarra, España.
lecharte@unav.es
FECHA DE RECEPCION: 2015-01-30
FECHA DE ENVÍO A PARES: 2015-01-30
FECHA DE APROBACIÓN POR PARES: 2015-06-07
FECHA DE ACEPTACIÓN: 2015-06-26
PARA CITAR ESTE ARTÍCULO / TO REFERENCE THIS ARTICLE / PARA CITAR ESTE ARTIGO
Pardo-Caballos A, Echarte-Alonso LE. La formalización del consentimiento informado en investigación y la pérdida sentimental del paciente. pers. bioét. 2015; 19(2): 198-226. DOI: 10.5294/pebi.2015.19.2.3
|
RESUMEN El consentimiento informado en investigación se inscribe en la relación médico-paciente, que ha sufrido, y sigue sufriendo, importantes cambios: desde el planteamiento clásico, como relación de amistad, al planteamiento contemporáneo, como relación entre extraños (enemigos potenciales, siguiendo tesis ilustradas sobre la sociedad), que es regulada por normas éticas y legales. El principal objetivo de estas últimas es conseguir, mediante requisitos formales, que dicha relación no perjudique al paciente. Bajo esta luz, se examina y compara el consentimiento informado en las principales normativas internacionales y en la ley española. Especialmente, nos centramos en la hoja de información al paciente, la comprensión, la voluntariedad, la certificación y la comunicación de los resultados de la investigación. Se concluye con la observación de la seria limitación intrínseca de la que adolecen estas normativas, que impiden, más que favorecen, el trato adecuado al paciente. PALABRAS-CLAVE: relación médico-paciente, Ilustración medica, investigación clínica, consentimiento informado, formalización del consentimiento (Fuente: DeCS, Bireme). |
ABSTRACT Informed consent for research is part of the doctor-patient relationship, which has suffered and continues to suffer important changes. These range from the classic approach, such as friendship, to the contemporary approach, namely, a relationship between strangers (potential enemies, according to illustrated notions of society) regulated by ethical and legal standards. The primary objective of these standards is to ensure, through formal requirements, that the relationship does not harm the patient. With this perspective in mind, the study examines and compares informed consent in light of the most important international standards and under Spanish law. There is a particular focus on the patient information sheet, understanding, willingness, certification and communication of research results. The study concludes there are serious inherent constraints in these regulations that prevent rather than promote treating the patient properly. KEY WORDS: Doctor-patient relationship, medical illustration, clinical research, informed consent, formalization of consent (Source: DeCS, Bireme). |
RESUMO O consentimento informado em pesquisa está apoiado na relação médico-paciente, que sofreu, e continua sofrendo, importantes mudanças: da proposta clássica, como relação de amizade, à proposta contemporânea, como relação entre estranhos (inimigos potenciais, seguindo teses ilustradas sobre a sociedade), que é regulada por normas éticas e legais. O principal objetivo destas últimas é conseguir, mediante requisitos formais, que essa relação não prejudique o paciente. Sob esse prisma, examina e compara-se o consentimento informado nas principais normativas internacionais e na lei espanhola. Especialmente, centra-se no prontuário do paciente, na compreensão, na voluntariedade, na certificação e na comunicação dos resultados da pesquisa. Conclui-se com a observação da séria limitação intrínseca da qual sofrem essas normativas, que impedem, mais do que favorecem, o tratamento adequado do paciente. PALAVRAS-CHAVE: relação médico-paciente, ilustração médica, pesquisa clínica, consentimento informado, formalização do consentimento (Fonte: DeCS, Bireme). |
El consentimiento informado (CI) es hoy uno de los tópicos más mencionados en la atención clínica y en los estudios de bioética que tocan, de algún modo, la relación médico-enfermo. En investigación clínica, esta presencia es todavía mayor.
Cuando se examinan estudios sobre el consentimiento informado, es fácil encontrar una cierta desconexión con otros elementos de la atención clínica. En el texto que sigue plantearemos la relación médico-paciente, y sus exigencias naturales, como marco de la investigación clínica y como fundamento del consentimiento informado, así como la deriva contemporánea de esta relación; expondremos el desarrollo de la formalización del consentimiento en tiempos recientes; y, por último, apuntaremos una nota crítica a las formalizaciones vigentes, que permita recapacitar sobre la realidad del paciente concreto que es atendido en la labor de investigación.
DE LA AMISTAD CON EL PACIENTE A LA DESCONFIANZA PROTOCOLIZADA
La relación entre médico y paciente ha sido considerada clásicamente como una forma de amistad, en el sentido más amplio del término: una relación benevolente que establece vínculos humanos, distinta de las amistades normales, pues se da entre personas desiguales y especialmente para beneficio de una de ellas, el enfermo. En este apartado examinaremos la naturaleza de dicha relación y su variación actual; esto, junto con una visión general de la investigación clínica, servirá de marco para examinar la aparición moderna de protocolos éticos en la investigación.
LA RELACIÓN MÉDICO-PACIENTE
Es clásico plantear el comienzo de una relación profesional en medicina como resultado de la visita del paciente al médico en busca de ayuda para un problema que piensa derivado de una enfermedad de cualquier tipo. Este planteamiento básico nos sitúa ya en un contexto con dos parámetros: se trata de una relación entre dos personas, en que una, el médico, está gravada especialmente por el deber de atender, prolongación peculiar del deber de sociabilidad que tiene cualquier persona, y la otra, el enfermo, se encuentra en situación de especial dependencia.
Para llegar a un conocimiento adecuado del problema del paciente, se debe desarrollar un diálogo con este, en el que aparecen los elementos básicos que habrá que tener en cuenta para el tratamiento. El contenido de dicho diálogo con vistas a conseguir la curación (si es posible), es descrito con especial vivacidad en Platón.3 Por una parte, está la cuestión que llamaríamos técnica: conocer la enfermedad desde sus comienzos y según sus fundamentos naturales (científicos, diríamos hoy); por otra, el médico habla con el enfermo, amigos y familiares, para saber cómo es su vivir, pues la salud es la capacidad de vivir la vida, y el vivir es peculiar de cada persona (2).
Con esos dos datos en la mano, el médico ya puede plantearse los medios adecuados para la recuperación del paciente; en la época de Platón, como muchas de las medidas terapéuticas implicaban un cambio de modo de vida, la labor del médico, antes de indicar el remedio, debía consistir en convencer al paciente de que lo que le iba a proponer era lo más adecuado para su situación. Es lo que hoy día correspondería a lo que llamamos educación sanitaria, que se suele plantear ahora especialmente con fines preventivos; en esta línea, actualmente es muy frecuente también el interés de los pedagogos en los pacientes hospitalizados, especialmente con pacientes pediátricos graves, para llevar a cabo esa acción docente que Platón propone para el médico, pues estos enfermos tienen especiales dificultades en adaptarse a su situación, y hay que llevarlos a la salud de la mano, en una labor de docencia especializada. De esta manera, se lleva a la práctica lo que Platón planteaba para un buen médico: más que hacer la indicación (lo más desagradable de su intervención) y marcharse, debe animar al paciente a asumirla.4
Con estas pocas frases, Platón deja planteada la acción médica como conjunta: el paciente no es un extraño en la acción terapéutica del médico, sino que asume lo que el médico le dice y, ayudado por él, lo lleva a la práctica.
Previamente, además, el médico ha conocido las peculiaridades humanas del paciente, de modo que ha asumido particularidades que lo llevan a variar su consejo. Dicho de modo sencillo: el consejo médico no se refiere, sin más, a lo técnico o científico de la enfermedad,5 sino que el paciente puede "regatear"; la salud no es una situación corporal perfecta que sea la meta de la acción sanitaria: la meta es la situación corporal que permite vivir el vivir humano, con sus peculiaridades concretas, de las que el médico se tiene que hacer cargo (dejando ideales fisiológicos un tanto de lado) antes de proponer un tratamiento.
Modernamente, se tiende a ver este diálogo con el paciente antes de plantear el tratamiento como una manifestación de la información a los pacientes, o como un embrión del actual consentimiento informado (3), del que hablaremos a continuación. El propio texto y la mentalidad de la época no dejan mucho lugar a esa interpretación. Parece más lógico entender este diálogo como la comunicación humana, rica en matices, que permite el entendimiento mutuo en una situación de confianza, y posibilita la acción solidaria de médico y paciente.
LA RELACIÓN MÉDICO-PACIENTE MODERNA
Aunque los autores que difundieron la mentalidad ilustrada desde el siglo XVII tienen desarrollos diversos, hay algunas notas comunes que han calado en la sociedad, y que forman parte hoy de nuestro imaginario colectivo. Una de esas notas es el modo de entender las relaciones humanas en la sociedad: se ha pasado de concebir la sociabilidad como algo natural del hombre a concebir que se trata de una sociabilidad artificial, pactada entre sujetos originalmente no sociales.
El motivo profundo que se plantea para sustentar este vínculo artificial entre los hombres es variado: evitar la muerte violenta a manos de otros (Hobbes), poder disfrutar de las comodidades de la vida por el intercambio comercial (Locke), poder disfrutar de la libertad para los impulsos naturales (Rousseau), etc. En todos los casos, un interés individual lleva a renunciar a algunos bienes posibles para, por medio del pacto social, conquistar mayores beneficios. Dicho pacto social impone algunas restricciones individuales, que adoptan la forma de reglamentaciones legales externas;6 estas garantizan (en teoría) que la sociedad va a funcionar y a prestar utilidad a todos los que se asocien a ella.
En suma, según la modernidad, el hombre es naturalmente egoísta, y en toda relación humana busca su beneficio, y solo su beneficio, aunque, con la adecuada articulación entre egoísmos (legal y externa), se consigue un beneficio para todos.7 Aquí cabría hablar de las muchas consecuencias que se observan contemporáneamente, pero nos limitaremos al tema que nos ocupa: la información al paciente.
En una relación humana que se considere que se establece entre potenciales enemigos o competidores (siguiendo un paradigma típicamente ilustrado), siempre está presente el temor al engaño realizado por la otra parte. Según el planteamiento moderno, la solución para garantizar el beneficio mutuo, a pesar de que la tendencia sería el engaño del otro en aras de los intereses personales, consiste en una normativa que proteja a la parte más débil.8
La idea del consentimiento informado se entiende cabalmente en este marco moderno. Para este planteamiento, el médico y el paciente son extraños que buscan el interés propio: el médico busca una remuneración y el paciente busca la salud... o alguna otra cosa que la técnica médica pueda proporcionar. Para evitar el engaño del paciente, se establece la obligación de informar por parte del médico para que el paciente, autónomamente, elija entre las opciones disponibles que el profesional le muestre (el paciente es considerado, al modo ilustrado, individuo autónomo). Dicha obligación de informar en la atención clínica se reglamenta de modo cada vez más minucioso, llegando a obligar en Estados Unidos a la sesión, agotadora tanto para el médico como para el paciente, en que el médico debe informarle verbalmente de todos los extremos de una intervención que se salga de lo banal o que tenga cierta importancia.
LA CORRUPCIÓN DEL CONSENTIMIENTO INFORMADO
Ante este panorama, se está tentado inicialmente a pensar que esa obligación de informar en detalle beneficiaría al trato entre médico y paciente, mejorando o facilitando su comunicación; como vimos al hablar del planteamiento clásico, dicha comunicación es imprescindible para que el médico se haga cargo del modo de vida del paciente, y para que este asuma, como coactor, la puesta en práctica del tratamiento. Sin embargo, la realidad, tras suficientes años de práctica, se muestra muy distinta: el consentimiento informado formalizado ha corrompido el diálogo confiado entre médico y paciente. Veamos un par de muestras de dicha degeneración.
Por una parte, informar al paciente según los requisitos legales vigentes y ofrecerle unas opciones no es dialogar con él. Un paciente acude a su médico como una persona dependiente, necesitada de ayuda y consejo: lo más lejano a la autonomía que quepa suponer. Puede que el consentimiento informado le oferte la ayuda, los datos técnicos, pero le niega el consejo, pues deja la elección en el terreno del paciente, y el médico no quiere implicarse pues, si lo hace, sería legalmente responsable del sesgo que ha tomado la decisión clínica, y se enfrentaría con posibles represalias legales (6).9 La pregunta clásica: "Doctor, usted, en mi caso, ¿qué haría?",10 ahora tiene muchas dificultades para conseguir un consejo; si la nueva mentalidad se encuentra profundamente implantada, obtiene solo un menú de posibilidades. La formalización legal del consentimiento ha conseguido que se atienda mal a los pacientes cuando, en teoría, pretendía justo lo contrario.
Otra repercusión clara es la conversión del consentimiento informado en una actividad rutinaria, como cualquier otro protocolo técnico más; así, en las historias clínicas en hospitales de Estados Unidos se hace referencia a que se ha hecho PARQ (que corresponde a explicar el procedimiento, alternativas, riesgos e invitar al paciente a formular preguntas -questions-); y se ha acuñado el neologismo de "consentizar" (to consent) al paciente.11 Evidentemente, tras este sistema protocolizado rutinario late una defensa ante posibles querellas de los pacientes. Y también oculta algo de lo que podría saberse de ellos: el artículo recién mencionado refiere una querella en que la paciente litigaba por haber sufrido complicaciones de una angiografía, alegando que no se le ofertó una prueba menos invasiva (una resonancia magnética); pero, ¿era realmente necesario aducir eso en la querella legal, o que simplemente un quirófano le inspiraba mucho más respeto? El consentimiento informado no puede ver ese detalle, mientras que un diálogo no estructurado lo manifestaría enseguida. Realmente, seguir las normativas del CI no permite conocer a la persona del paciente-voluntario.
Todos estos problemas derivados de la formalización del consentimiento informado no han pasado inadvertidos, y han provocado un auge reciente de los métodos cualitativos y no estructurados para acercarse a la realidad del enfermar, o a las cuestiones éticas de la práctica médica; concretamente, además de las entrevistas abiertas a pacientes y médicos, ha crecido en gran medida la importancia de las narrativas en estos contextos. Estos procedimientos, al no encasillar la realidad, no la recortan, y permiten hacerse cargo más cabalmente de lo que está pasando o, al menos, de algunas facetas no recogidas en los protocolos o las normativas (que no dejan por eso de tener utilidad).
LA INVESTIGACIÓN CLÍNICA
Para entender adecuadamente la investigación clínica, es necesario enmarcarla dentro de la atención a los pacientes. Investigar, en este contexto, es proponer a los pacientes un tratamiento novedoso, insuficientemente probado en ese momento, que quizá pueda serle de ayuda cuando otras medidas terapéuticas probadas se muestran insuficientes, o incluso solamente prometen alguna ventaja ligera con respecto al tratamiento, eficaz, que venía recibiendo. Esta peculiaridad de la investigación no quita nada a la relación médico-paciente: persiste la obligación de diálogo para conocer sus particularidades humanas y aprender de él, y para ayudarle a hacerse cargo de su situación y facilitarle su misión de llevar adelante el tratamiento junto con quien le cuida. Estos elementos constituyen así la base de los requisitos éticos de la investigación; tienen cierto parecido con la obligación de informar al paciente y la condición de que este aporte su consentimiento, pero son muy distintos y mucho más amplios.
En este sentido, se observa con frecuencia un enfoque de la ética de la investigación que la plantea como si esta fuera una empresa "independiente" o "autónoma", con leyes propias, y la ética se dedicara a poner barreras a la ciencia para proteger a los sujetos de investigación; como si la investigación tuviera poco o nada que ver con el tratamiento de los pacientes. El resultado es la formalización de las reglas de la investigación, especialmente del consentimiento informado: la investigación se ve forzada a implicar a los enfermos en su sistema de desarrollo de pruebas diagnósticas o tratamientos, y entonces busca su consentimiento. Más adelante veremos con detalle los factores que contribuyen a la formalización del consentimiento en investigación.
ORIGEN DE LA FORMALIZACIÓN DEL CONSENTIMIENTO EN INVESTIGACIÓN
Aunque luego se explica pormenorizadamente el desarrollo histórico de las normativas de la investigación clínica, haremos aquí un apunte para ver cómo se inició y desarrolló su formalización que, como acabamos de decir, intenta encasillar cuestiones básicas de la relación médico-paciente cuando el médico propone algo no suficientemente probado todavía.
El inicio viene marcado por el juicio de Nüremberg a los criminales de guerra alemanes de la Segunda Guerra Mundial; este juicio se encontró con carencias serias de reglamentaciones específicas sobre investigación médica que sirvieran de fundamento para poder condenar a los culpables. Por este motivo, el tribunal elaboró una normativa muy sencilla, llena de lógica, para la investigación clínica,12 que sirviera de referencia para sus sentencias. El documento entero cabe en una página sin apreturas.
Debido a los abusos realizados por la medicina nazi, el acento de este documento se carga sobre el consentimiento informado, apartado mucho más extenso que cualquiera de los demás. Sin embargo, se observa en los puntos 9 y 10 una referencia a la sensatez tanto del paciente como del médico a la hora de detener la investigación que esté causando problemas.13 El paciente debe plantear el fin de la experimentación si ve que las molestias o los problemas derivados del experimento se le hacen incomportables, frenando un posible entusiasmo del médico, y otro tanto debe hacer el médico, frenando el entusiasmo del paciente. Se ve aquí reflejada, indirectamente, una relación médico-paciente concebida al modo clásico, como un entendimiento mutuo y una acción conjunta, regulada por la prudencia de ambos actores.
La historia posterior del "código" de Nüremberg es un tanto penosa:14 el fenómeno psicológico que lleva a pensar que "los buenos" hemos ganado la guerra, creó el imaginario colectivo de que los abusos médicos durante la guerra, e inmediatamente antes, habían corrido a cargo de médicos alemanes exclusivamente. Se olvidó que los abogados en Nüremberg, en la defensa de los acusados, aportaron como justificación datos de investigaciones similares, inadmisibles, que habían tenido lugar en los países aliados en ese mismo periodo. El hecho es que el texto de Nüremberg no tuvo derivaciones y enriquecimientos en textos posteriores, sino que quedó como una anécdota histórica aislada: solo dos décadas después aparecieron nuevas reglamentaciones o normativas relativas a la ética de investigación, como puede verse en las anotaciones de la versión inglesa del Informe Belmont.15 Veamos ahora esas nuevas normativas.
PROGRESO DE LA FORMALIZACIÓN Y CAMBIO DE PARADIGMA
No es necesario mostrar con mucho detalle que la investigación éticamente incorrecta se encuentra en cualquier periodo de la historia de la medicina, y especialmente en épocas modernas, con el método científico más desarrollado. El periodo posterior a la Segunda Guerra Mundial no ha sido una excepción. En los años sesenta fueron apareciendo en los medios, tanto generales como profesionales, noticias de escándalos en investigación que movieron a elaborar normativas que detuvieran esas actividades que se estaban conociendo. Destacan entre ellos el ya mencionado informe Belmont, de 1978, y la Declaración de Helsinki de la Asociación Médica Mundial,16 de 1964 que, por el contexto de la época, pueden considerarse contemporáneos. A diferencia del texto de Nüremberg, muy sencillo, ambos documentos muestran una formalización más prolija.
El informe Belmont, elaborado en Estados Unidos, hace una referencia inicial al texto de Nüremberg y se extiende con más detalles sobre los principios éticos básicos que se deben tener en cuenta (respeto a las personas, beneficencia y equidad), y sus aplicaciones (consentimiento informado —que debe incluir información, comprensión y voluntariedad—, valoración de riesgos y beneficios —y aparece la figura de un comité de evaluación ya exigida por otras normativas estadounidenses— y la justicia en la selección de sujetos de investigación). Este texto puede considerarse, por su planteamiento, como una extensión del de Nüremberg.
La Declaración de Helsinki, sin embargo, toma un rumbo muy distinto, aunque en apariencia similar. Por supuesto, plantea correctamente los fines de la investigación, la protección del sujeto de investigación, el balance daño-beneficio, etc.: son los elementos parecidos (si los viéramos en detalle, observaríamos que tienen también sus diferencias). Pero surgen dos cuestiones nuevas: la investigación prevista debe ser revisada y aprobada por un comité de ética de investigación que sea independiente del médico o el equipo investigador (punto 23 de la revisión vigente), y el paciente que participa en la investigación puede retirarse de ella cuando quiera sin exponerse por ello a represalias (punto 26 de la revisión vigente). Con estos dos requisitos, pasamos de un contexto de relación médico-paciente clásica de confianza, entendimiento y responsabilidad mutua, a un contexto de relación entre individuos autónomos y extraños regulado por normativas e instituciones externas. Ya no se apela a la conciencia del paciente ni a la del médico: habrá un comité que supervise lo que quiere hacer el médico (con la suposición de que no cabe esperar de él nada bueno, añado, y por eso hay que fiscalizarlo), y el paciente pasa a ser un sujeto autónomo, al que no liga ningún deber, y que puede dejar la relación clínica y de investigación que ha emprendido, simplemente porque se va de vacaciones (entre otros escenarios posibles, y a pesar de los perjuicios que pueda ocasionar).
Tenemos así dibujado el marco de la situación actual: parece darse por supuesto que la investigación clínica no es una extensión de la atención al paciente, sino una actividad independiente, que se realiza entre extraños; para regularla y conseguir que se desarrolle en beneficio de todos, se elaboran las formalizaciones normativas que ya hemos esbozado y que veremos en detalle más adelante.
FACTORES QUE CONTRIBUYEN A LA FORMALIZACIÓN
El nuevo paradigma de la investigación ha seguido la huella de la filosofía política moderna, como tantas otras cuestiones en la sociedad actual. A su vez, los teóricos ilustrados que desarrollaron esas ideas eran herederos de su mentalidad y su modo de vivir, que se pueden rastrear en sus teorías. Dicho de otro modo: las maneras de enfocar las cuestiones tienen mucho que ver con el modo de pensar y vivir. Con lo que se nos plantea una última pregunta: ¿por qué esta deriva contemporánea de la ética de la investigación a una normativización con control externo, en detrimento de una relación de confianza entre médico y paciente? Aquí, nos limitaremos a apuntar una idea de fondo y dos cuestiones relativas al imaginario colectivo actual. En todas ellas está latiendo el ideal del desarrollo científico moderno.
Es bien sabido que el moderno método científico pretendía, desde sus orígenes, elaborar una nueva ciencia, cuyo objetivo no sería saber, sino poder transformar la realidad para ponerla al servicio del hombre; esto ya se encuentra en Bacon, y es más claro todavía en Hobbes: el objetivo es poder hacer cosas (algo que la filosofía nunca pudo ni podrá). Comte, en el siglo XIX, coherentemente con esta idea de partida, lleva la cuestión a sus últimas consecuencias: también la sociedad debe ser estudiada con el método científico (es el comienzo de la sociología positiva moderna), para poder actuar sobre ella de modo científico, de manera que se consiga, mediante la manipulación técnica, que alcance los objetivos que nos hemos fijado: la felicidad para todos, que se plantea para el futuro. El progreso (idea típica del siglo XIX) nos conducirá a ese estado de cosas ideal. No hay que insistir mucho en que, de hecho, cuando se llevó a la práctica, nos condujo en el siglo XX a algunas de las situaciones más espantosas que ha conocido la humanidad: dictaduras, genocidios, guerras mundiales y otros sucesos con millones de muertos a sus espaldas; una presunta felicidad en este mundo que, aparte de no conseguirse, ha resultado muy cara de pagar. En todo caso, a pesar de toda esta historia aciaga, la idea de partida persiste impávida, apoyada por sus indudables éxitos (que sería injusto negar): la técnica es buena, y —extrapolación excesiva— fomentarla solo nos llevará a cosas buenas y deseables.
En este contexto de predominio de la técnica, pronto se ve que se puede aplicar sobre el propio hombre, que pasa a ser materia para esa transformación. Esto abre nuevos horizontes a los deseos del ser humano: ya no se tienen que limitar a la posesión de objetos externos, sino que pueden optar por modificar su propio cuerpo a voluntad; toda la discusión sobre el transhumanismo descansa sobre esta idea; pero, de modo más cercano, ya se ha llevado a la práctica con la difusión de la contracepción o las técnicas de reproducción in vitro: si yo quiero o no quiero hijos ahora, tengo a la técnica como aliada para conseguir lo que quiero cuando lo quiero. Sin embargo, Lewis apunta con agudeza (13) que el progreso técnico al servicio de los deseos del hombre, especialmente los aplicados al cuerpo humano, se convierte, de hecho, en el progreso del dominio de los hombres que poseen las técnicas sobre los que no las poseen, que se ven así manipulados según el capricho de los que las tienen. Así, el hombre pasa a ser, de beneficiario de la transformación técnica de las cosas, a materia para dicha transformación, según los deseos de otros. La investigación científica sobre seres humanos es la muestra paradigmática de esta manipulación o uso de otra persona para fines ajenos. La reacción lógica es la defensa. Y esta, en la civilización técnica, se realiza mediante una técnica: las normativas que regulan ("científicamente") la sociedad, e impiden que otros miembros de esta inculquen el ámbito de mi autonomía. La formalización normativa del consentimiento informado en investigación es lo que cualquier persona actual pensaría en este contexto.
Hemos mencionado la actitud defensiva del paciente: ante la incertidumbre que va aneja a toda investigación, la normativa le aporta protección y sensación de seguridad. Sin embargo, también el médico se encuentra en una cierta situación insegura, que ya hemos mencionado a propósito de la atención clínica normal: siempre está la espada de Damocles de una querella por parte del paciente. La normativa de investigación también juega a su favor: si se cumple lo que está mandado, el médico tiene la tranquilidad de que no podrá ser acusado de mala práctica. La reglamentación de la investigación, por tanto, resulta un refugio adecuado para todos los actores, a pesar de las deficiencias que pueda comportar. Por último, la práctica de la atención médica es cada vez más compleja; los contenidos técnicos de la medicina se han multiplicado en tiempos recientes. Desde un punto de vista meramente técnico, la medicina se ha vuelto cada vez más difícil de abarcar. Para una buena práctica clínica, se hacen necesarias guías de actuación técnica. Aunque el razonamiento del médico sobre el caso concreto del paciente que atiende es insustituible, los protocolos de actuación técnica, y los diagramas de flujo para el diagnóstico o la toma de decisiones terapéuticas, son herramientas útiles, si se saben utilizar. El médico, en muchas ocasiones, piensa que no tiene más que dejarse guiar por estas indicaciones para llegar a un buen resultado. Esta actitud resulta empobrecedora, en cuanto que limita el razonamiento sobre el caso concreto que se tiene entre manos. Por este motivo, los protocolos y las guías de actuación son interesantes y orientadores, pero hay que conservar siempre el espíritu crítico para poder llegar a una aplicación más específica para el paciente que estamos atendiendo, siempre con peculiaridades que difícilmente están contempladas en un protocolo o guía.
Esta costumbre —y quizá cierta pereza—, de confiar en protocolos termina extendiéndose a la investigación: aunque se pueden razonar muchas cuestiones concretas en nuestra relación con el sujeto de investigación, seguir un protocolo parece al médico un procedimiento seguro para no omitir nada digno de mención. Por último, la formalización del consentimiento en investigación, que viene ya impulsada por otras vías, se ve reforzada por el hábito del médico de fiarse de protocolos y guías de actuación.
LA FORMALIZACIÓN DEL CONSENTIMIENTO INFORMADO
El consentimiento informado como herramienta normalizada en la investigación es la declaración expresa del voluntario que manifiesta haber entendido cuál va a ser su participación y en la que se deja constancia de su libre colaboración. Tiene como finalidad proteger a todos los participantes en el ensayo —tanto a voluntarios e investigadores— en su dignidad, derechos y bienestar. En este apartado vamos a estudiar su evolución histórica, los elementos éticos que lo integran, las características de algunos modelos especiales así como las circunstancias que eximen de este requisito formal.
MARCO HISTÓRICO Y LEGAL
Entre los primeros antecedentes de regulación del consentimiento informado (CI) en la investigación se encuentra la sentencia de la Corte Inglesa de 1767 (caso Slater vs. Baker & Stapleton) a favor de un paciente al que dos médicos habían desunido experimentalmente una fractura parcialmente consolidada sin su consentimiento (14). Hay que destacar que la sentencia ya refleja la necesidad legal del CI basándose en una razón y un hecho. La razón se refiere al ataque a la persona que supone toda manipulación del cuerpo sin consentimiento. Pero, lo que es más interesante, la sentencia recoge como hecho la costumbre entre los cirujanos ingleses de no utilizar nuevas técnicas potencialmente terapéuticas sin la autorización del paciente.
Ciertamente, si bien el proceso de CI se formaliza posteriormente al siglo XVIII, esta sentencia muestra que, en cierta manera, su obtención se ha tenido en cuenta a lo largo de la tradición investigadora médica. Hay que añadir que, sin embargo, en la mayoría de las ocasiones, se trataba de un consentimiento de mínimos. Es decir, no implicaba revelar apenas información sobre las pruebas experimentales realizadas. De hecho, en los siglos XVIII y XIX, la mayor parte de la literatura científica sobre investigación justifica incluso la distorsión de los datos y las expectativas para motivar la colaboración de los pacientes.
A finales del siglo XIX encontramos ya el primer incremento significativo de testimonios sobre solicitudes sistemáticas y formalizadas del consentimiento, tal como hoy lo conocemos. Uno de los casos más notorios es el relacionado con las primeras inoculaciones de la tuberculina. Los médicos estadounidenses que se ocuparon de probar este tratamiento incluyeron, como parte de sus protocolos, la obligación de informar al colaborador antes de obtener su permiso (15).
A principio del siglo XX cabe mencionar a Walter Reed como uno de los más notables pioneros en el uso del CI (16). También todos los voluntarios que participaron en sus indagaciones sobre la fiebre amarilla firmaron un documento de libre colaboración. Por esas mismas fechas, el Gobierno alemán redacta las primeras leyes reguladoras de la investigación médica. Estas surgen como respuesta al escándalo ocasionado por los experimentos de Albert Neisser sobre el estudio de la sífilis en pacientes expresamente contagiados. Como contrapartida, en 1902, la American Medical Association rechaza admitir la recomendación de incluir la solicitud del CI en los protocolos de investigación. No será hasta 1935 cuando la Corte Americana reconozca explícitamente tal necesidad (17).
Hay que esperar hasta el final de la Segunda Guerra Mundial para que se redacten los primeros esbozos de regulación internacional acerca del CI en la investigación biomédica. En los diez puntos del Código de Nüremberg (1947) aparecen ya alusiones explícitas sobre este tema:
...el consentimiento voluntario del sujeto humano es absolutamente esencial. Esto quiere decir que la persona afectada deberá tener capacidad legal para consentir; deberá estar en situación tal que pueda ejercer plena libertad de elección, sin impedimento alguno de fuerza, fraude, engaño, intimidación, promesa o cualquier otra forma de coacción o amenaza; y deberá tener información y conocimiento suficientes (18).
A estos tres requisitos el Código de Nüremberg añade una cuarta y fundamental exigencia referente a los compromisos del grupo investigador: "El deber y la responsabilidad de evaluar la calidad del consentimiento corren de la cuenta de todos y cada uno de los individuos que inician o dirigen el experimento o que colaboran en él. Es un deber y una responsabilidad personal que no puede ser impunemente delegada en otro". Es revelador que este último punto, que pretende cerrar las puertas a los intentos de subrogación de delitos hacia cargos superiores —ya sean de carácter civil o militar—, apunte específicamente a la obtención del consentimiento.
Posteriormente, se incorporó la necesidad de obtener el CI en la Declaración de Helsinki, elaborada por la Asociación Médica Mundial (AMM) en 1964. Para los casos de incapacidad legal, la AMM introduce la figura del representante legal como intermediario entre el paciente y el investigador en el proceso de obtención del CI (19).
Henry Beecher publica en 1966 un artículo (20) en que denuncia 22 investigaciones dentro de Estados Unidos que contravenían de un modo u otro las reglas éticas de Helsinki, y fuerza así la implantación ese mismo año por parte del Public Health Service de una normativa más restrictiva respecto a la exigencia del consentimiento. Más tarde, el caso Tuskegee (21), precipita, en 1974, la creación de la Comisión Nacional para la Protección de los Sujetos Humanos frente a la Investigación Biomédica y de la Conducta. Sus componentes darán lugar, cuatro años después, al Informe Belmont (22). En lo que al CI concierne, la mayor aportación del Informe Belmont está relacionada con "la forma y el contexto" en el que se ha de ofrecer la información al voluntario. "Dado que la capacidad para entender es función de la inteligencia, la racionalidad, la madurez y el lenguaje, es necesario adaptar la presentación de la información a las capacidades del sujeto". Tareas que, según el Informe Belmont, competen directamente al equipo médico: "los investigadores son responsables de averiguar si los sujetos han comprendido la información".
Con el Informe Belmont de trasfondo, se han realizado varias revisiones de la Declaración de Helsinki en sucesivas asambleas de la AMM. La efectuada en Venecia en 1983, está especialmente relacionada con el CI relativo a las investigaciones sobre grupos vulnerables. Entre ellos reconoce a "menores, prisioneros, mujeres embarazadas, fetos humanos o adultos física o mentalmente incapacitados para dar su consentimiento". Además del consabido consentimiento del representante legal, se recomienda lograr también el asentimiento del menor. Por otro lado, en esta enmienda se hace alusión además al problema de pacientes que no pueden dar su consentimiento directo o indirecto (por representante legal). En estos casos, se estipula que los investigadores solo puedan incluir su participación "si la condición física/mental que impide obtener el consentimiento informado es una característica necesaria de la población investigada". En todo caso, la justificación debe ser siempre incluida en el protocolo de evaluación para la aceptación del proyecto y bajo la condición de obtener, en la mayor brevedad posible, el CI del individuo o de un representante legal (Principio 1°. 11, 1983).
ACTUALES DOCUMENTOS DE REFERENCIA
La guía ética de mayor reconocimiento internacional es la promovida por la Asociación Médica Mundial, que acabamos de comentar. Tuvo su última modificación en la asamblea de Fortaleza (Brasil), en 2013.
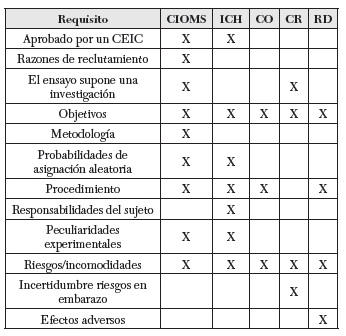
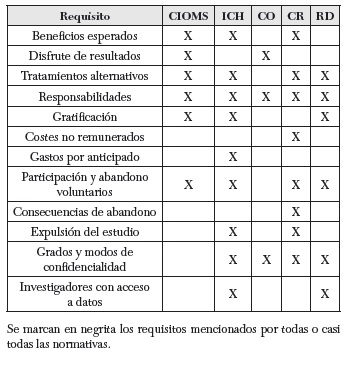
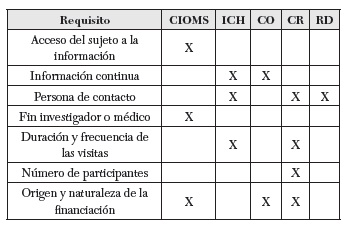
También es muy conocida y utilizada la International Guidelines for Biomedical Research Involving Human Subjects redactada por el Council for International Organizations of Medical Sciences (CIOMS) (23). Un apartado destacable en lo que respecta al CI es su directiva quinta, donde podemos encontrar una lista de los contenidos esenciales de la Hoja de información al paciente (HIP), un documento en el que aparece toda la información comunicada a este antes de la obtención su consentimiento. Por otro lado, en su directiva séptima se encuentran claramente reflejados los cuatro elementos que hoy se consideran claves para el reclutamiento de voluntarios (este punto será desarrollado específicamente en un apartado posterior).
Otro texto de referencia es la Guía de Buenas Prácticas Clínicas (ICH Harmonised Tripartite Guideline) de la Conferencia Internacional de Armonización (24). Desde 1996, provee un modelo unificado para la Unión Europea, Estados Unidos y Japón sobre la realización de estudios clínicos. Relacionado con el CI cabe mencionar una HIP más sintética que la de la CIOMS — solo 20 puntos respecto a los 26 de la primera—; cinco condiciones eximentes de obtención del CI; y varias recomendaciones respecto a situaciones de emergencia. Todos estos puntos también serán comentados más detenidamente más adelante.
Otro documento destacable internacionalmente pero, además, con peso jurídico en Estados Unidos, es la llamada Common Rule (25), aprobada como ley federal en 1991. Sus dos principales puntos de regulación giran en torno al requerimiento del CI y al requisito de aprobación por parte del Institutional Review Board (IRB) de toda investigación con seres humanos hecha dentro de Estados Unidos. Es interesante remarcar que la lista que se ofrece acerca de la información sobre la HIP es tomada como modelo de aplicación en la mayor parte de los proyectos de investigación de índole internacional.
Centrándonos en la Unión Europea, hay que mencionar la normativa del Additional Protocol to the Convention of Human Rights and Biomedicine Concerning Biomedical Research (26), enmarcado dentro del Convenio de Oviedo. Encontramos en este documento la hoja de información del CI más escueta, con tan solo ocho puntos. A pesar de ello, es el texto en el que se hace mayor hincapié sobre el modo de protección de los sujetos incapaces a la hora de dar su consentimiento, así como en la protección de embarazadas, lactantes y presos. Por otra parte, la brevedad de este documento pretende facilitar su manejabilidad, lo que le otorga un especial valor. A esto hay que añadir la existencia de un documento explicativo, anexo, en el que se desarrollan las cuestiones de carácter más conflictivo o ambiguo. Por último, y como contenido también novedoso, encontramos en los apéndices una mención expresa al deber de los investigadores de informar sobre el modo como se ha dado la información al voluntario, dentro del informe del proyecto para su evaluación por parte de los comités éticos (punto xii).
Además de servir de punto de apoyo para la comunidad científica internacional, las directrices mencionadas han sido implementadas como norma jurídica en muchos países, confiriéndoles con ello un especial peso legal. Uno de estos casos es el de España, que comienza a regular la investigación en 1978 con el Real Decreto de Ensayos Clínicos (27); posteriormente, con la Ley del Medicamento de 1990 (28); el Real Decreto 561/1993 (29); y, finalmente, con el Real Decreto 223/2004 (30), que actualiza el del año 1978, y la ley 14/2007 (31). Concretamente, el primer punto del artículo 7 —donde este último Real Decreto desarrolla específicamente la regulación en torno al CI— reconoce la deuda y el valor interpretativo que aportan las recomendaciones europeas, especialmente las del Convenio de Oviedo. Por último, y dada la importancia que tiene el Real Decreto 223/2004 de cara a la investigación llevada a cabo dentro de España, haremos especial referencia a él a partir de ahora, quedando simplemente presentado en este epígrafe.
Para terminar, hemos recogido en la siguiente tabla los contenidos que, según algunas normativas nacionales e internacionales, debieran ser incluidos en las hojas informativas del proceso de CI. Las normativas seleccionadas son: a) las pautas éticas internacionales para la investigación biomédica en seres humanos del Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS); la Guía tripartita y armonizada de la buena práctica clínica (ICH); el protocolo de investigación surgido en el 2005 en el marco del Convenio de Oviedo (CO); los recogidos en el Código de Reglamentos Oficiales norteamericano de 1991, también llamada Regla Común (CR) y, por último, los regulados en España con el Real Decreto sobre regulación de ensayos clínicos, aprobado en el 2004 (RD):
En los subapartados siguientes examinaremos con más detalle los requisitos éticos del CI, tal como vienen recogidos en estas normativas.
RECLUTAMIENTO DE LOS PARTICIPANTES
La participación en un estudio clínico siempre conlleva molestias y riesgos que, en mayor o menor medida, pueden suponer motivos para declinar la invitación a colaborar en él. Incluso, investigaciones que requieran solo tareas sencillas, como rellenar encuestas, pueden inquietar y afectar de manera negativa a los sujetos de estudio. Por esto, un requisito previo a toda posible colaboración es siempre informar sobre qué tipo de repercusiones puede tener el estudio en el participante. Tales inconvenientes son compensados, en algunos casos, por el potencial beneficio directo que puede suponer la colaboración del voluntario. A este respecto, el Real Decreto 223/2004 establece que "el sujeto o sus representantes legales únicamente podrán recibir del promotor el reintegro de los gastos extraordinarios y pérdidas de productividad que se deriven de su participación en el ensayo" (art. 3, punto 9).
No obstante, en los estudios sin un beneficio potencial directo para el sujeto colaborador, puede ofrecerse una compensación económica para incentivar el reclutamiento. Pero estas deben estar en relación con "las características del ensayo y, en ningún caso, tan elevadas como para inducir al sujeto a participar por motivos distintos del interés del avance científico" (Real Decreto 223/2004, art. 3, punto 8). Esta precaución es subrayada con mayor énfasis en la Guidance for Institutional Review Boards and Clinical Investigators de la Food and Drug Administration (FDA). En ella se establece que, como el pago a sujetos de investigación no debe considerarse un beneficio real, no tiene que incluirse como tal en la lista de beneficios de participación de la hoja de información, sino en una sección aparte.
Un caso extraordinario de compensación por reclutamiento es el de los menores de edad e incapaces. El Real Decreto protege a dichos participantes de toda explotación mediante la prohibición de cualquier compensación económica "a excepción de los gastos extraordinarios y pérdida de productividad que se deriven de la participación del sujeto en el ensayo" (art. 3, punto 8).
Hay que valorar otra circunstancia importante con respecto al reclutamiento de sujetos para estudio: la condición del "paciente voluntario"; este es un enfermo que acude o está ingresado en un centro médico y que, a su vez, es invitado a colaborar en un ensayo clínico. En estos casos, es frecuente que sea el médico responsable del paciente quien ofrezca la invitación para la colaboración. Esta es una situación peculiar pues, al ser la relación médico-paciente de carácter fiduciario, es fácil que se produzca cierta coerción, aun de carácter inconsciente, sobre el potencial voluntario. Por esto, el médico debe mantener especial sensibilidad y precaución respecto a las invitaciones en su consulta, distinguiendo previamente en qué pacientes realmente procede solicitar su colaboración y sobre cuáles hay que evitar innecesarias molestias por su especial vulnerabilidad. Porque, en definitiva, toda estrategia de reclutamiento en centros asistenciales ha de evitar que la investigación biomédica afecte a la relación terapéutica. En todo caso, la utilización de incentivos económicos solo es admisible para el reclutamiento de sujetos sanos, nunca de pacientes.
ELEMENTOS ÉTICOS DEL CI
La definición de CI del artículo 2 del Real Decreto 223/2004 —artículo en el que se acotan los conceptos utilizados en la regulación de ensayos clínicos—, queda fijada en los siguientes términos: "decisión, que debe figurar por escrito y estar fechada y firmada, de participar en un ensayo clínico adoptada voluntariamente por una persona capaz de dar su consentimiento tras haber sido debidamente informada y documentada acerca de su naturaleza, importancia, implicaciones y riesgos".
A pesar de su formulación como imperativo legal, la obtención del CI no es un mero requisito burocrático, sino un proceso esencial en la ética de una investigación. Empieza antes de que el sujeto dé el consentimiento y continúa mientras esté comprometido con la investigación o le sea pertinente la información o los resultados relativos a su participación. Pero, pese a la importancia que suponen los elementos éticos del CI para toda investigación, no existe todavía hoy una directiva clara en España sobre ellos. Y si bien el Real Decreto recoge algunos de los puntos más importantes, faltan otros, aparte de no proporcionar las suficientes especificaciones como para evitar ambigüedades en algunas de las indicaciones. Un ejemplo más logrado de documento regulador es el proporcionado por un comité de Estados Unidos, el Western Institutional Review Board (WIRB) (32), que merece la pena mencionar aquí, ya que es utilizado como referencia en muchos otros países y empleado como modelo en numerosos protocolos internacionales de investigación. En este documento se señala a los investigadores que en el proceso de CI deben perseguirse los siete objetivos siguientes:
Dar al sujeto la información sobre la investigación.
Asegurarse de que el sujeto tiene tiempo suficiente para considerar las opciones.
Responder todas las preguntas del sujeto antes de que tome la decisión.
Comprobar que el sujeto ha entendido toda la información.
Obtener el consentimiento informado voluntario del sujeto que va a participar.
Continuar informando al sujeto a lo largo del estudio.
Continuar revalidando el consentimiento del sujeto durante toda la investigación.
Es posible distinguir en dicho proceso cuatro pasos fundamentales, que permiten ordenar los elementos éticos del CI:
El reclutamiento de participantes.
La información sobre la participación, los beneficios y riesgos del estudio.
La voluntariedad del paciente a lo largo del estudio.
El destino de las muestras y la información acumuladas.
En este texto vamos a desarrollar los primeros tres puntos, dejando aparte el cuarto dadas sus peculiaridades, que lo apartan en buena medida de la relación clínica con el paciente.
LA HOJA DE INFORMACIÓN AL PACIENTE
La libre aceptación en la participación en un ensayo clínico queda sujeta a que el potencial colaborador manifieste que haya recibido y comprendido toda la información relevante en la toma de decisiones. El punto 2, artículo 7, del Real Decreto 223/2004, hace hincapié en que la hoja de información tenga solo los datos relevantes (objetivos, riesgos, inconvenientes y el derecho a abandonar el ensayo), expresados en términos claros y comprensibles para el sujeto.
Información y comprensión son dos aspectos de una misma realidad comunicativa establecida entre el investigador (emisor) y el voluntario (receptor). El acto locutivo del investigador, realizado mediante la entrevista personal y la entrega de la hoja informativa, no cumple con su finalidad si el paciente no comprende la información facilitada para actuar en consecuencia (acto ilocutivo). A la inversa, el acto ilocutivo, que supone el consentimiento del voluntario tras la adecuada comprensión de los riesgos y beneficios de la participación en el estudio, no es legítimo si la información facilitada no es suficiente. En ambos casos, el acto comunicativo que supone la firma de un CI sería ineficiente.
Según el WIRB, la información facilitada en el proceso del CI debe comprender ocho apartados. El primero se refiere a la naturaleza intrínsecamente abierta de la hoja informativa. El investigador tiene que expresar la posibilidad de que el potencial participante pueda realizar libremente cualquier clase de preguntas a los responsables del proyecto, de llevarse el texto a casa para valorarlo con tiempo o para pedir una segunda opinión a expertos no involucrados en el estudio. Además, ha de señalar el derecho del paciente a rehusar la invitación o retirarse de la investigación en cualquier momento.
El segundo apartado está relacionado con el propósito del estudio. Aquí pueden incluirse datos como su duración, número de participantes y las evaluaciones previas a las que ha sido sometido su protocolo. En este punto, es importante añadir el carácter "experimental" del estudio. En otras palabras, el colaborador debe entender que no hay plenas garantías sobre sus efectos y resultados. Por último, hay que hacer referencia a la razón por la que el sujeto ha sido invitado a participar. Por ejemplo, "Debido a que usted ha sido diagnosticado de...".
El tercer apartado versa sobre los procedimientos del estudio de acuerdo con el protocolo: las técnicas empleadas, aquellas de carácter más experimental, el número de visitas, etc. Es importante mencionar el método por el cual se determina el grupo placebo y la probabilidad de ser asignado a dicho grupo. Además, en esta sección corresponde advertir de las actividades, viajes, hábitos, consumo de fármacos, alimentos, etc., a los que el voluntario deberá renunciar durante el estudio.
El siguiente apartado describe los riesgos de la participación en el estudio. Es importante señalar su probabilidad en la medida de lo posible, desestimando aquellos no relevantes o improbables. A esto hay que añadir la mención de las molestias derivadas de las técnicas invasivas (extracción de sangre, biopsias, intubación, etc.) y sobre la posibilidad de aparición de efectos adversos desconocidos. En el caso de embarazo debe informarse de los riesgos al feto. También, si el voluntario es un paciente sometido a tratamiento experimental, es necesario prevenir sobre la posibilidad de empeoramiento en el estudio. En último lugar, hay que nombrar los seguros que asisten al participante y quién asume las responsabilidades de los daños.
El quinto apartado trata de la estimación de beneficios para el voluntario, siempre indicando que estos no están garantizados. Pueden añadirse también aquí los potenciales beneficios sociales o para futuros pacientes, aunque siempre utilizando con cautela este tipo de previsiones. El disfrute de procedimientos, técnicas diagnósticas, tratamientos, etc., puede ser incluido aunque sin relacionarlo con el "coste" que acarrearían estos fuera del estudio, pues ello supondría considerarlo como una forma de pago. De nuevo, tal como se ha comentado, la mención a todo "pago indirecto" debe detallarse fuera del apartado de beneficios de la HIP, ya que esa es una manera de reflejar que dicha remuneración no es el determinante del sujeto que va a participar.
El sexto y el séptimo apartados se refieren a las alternativas y al coste. Primero, si rechaza participar, qué opciones tiene el paciente dentro y fuera del estudio experimental, así como, resumidamente, el riesgo y beneficio de dichas alternativas; y segundo, el gasto que va a suponer la participación del sujeto en la investigación y el nombre del investigador o la compañía responsable de asumir parcial o totalmente dichos costes. Sobre este asunto es interesante detallar, para evitar posteriores conflictos, la lista de gastos que van a ser sufragados y su modo de pago.
El último apartado mencionado por el WIRB es el de los incentivos de reclutamiento (directos o indirectos): si los hay, y el modo de pago (en sucesivas cuotas, según las visitas realizadas durante el estudio, al final del estudio, etc.). Es conveniente señalar, además, qué proporción de incentivo obtendría el voluntario si abandonase el ensayo, según en qué fase de la investigación y por qué motivos.
A estos ocho apartados es posible añadir uno más, enfatizado especialmente en el Convenio de Oviedo: hay que garantizar al paciente la confidencialidad en lo que a información sensible se trata. El futuro participante debe saber que todo dato obtenido en el ensayo clínico que pueda perjudicarlo estará suficientemente protegido. Por ejemplo, en el proceso de reclutamiento de participantes es frecuente exigir pruebas de VIH. Los resultados de este tipo de pruebas han de ser encriptados cuidadosamente y es necesario informar de dichos procedimientos.
LA COMPRENSIÓN
Como se mencionó al principio del apartado, conocer los contenidos de la hoja informativa no basta para llevar acabo eficazmente un proceso de CI. Hace falta saber también cómo ofrecer dicha información. Ello dependerá del tipo de estudio y de las características del participante.
Respecto al tipo de estudio, hay que evitar caer en el error de pensar que el modo comunicativo es idéntico al de la interacción médico-paciente. Este criterio es inadecuado, pues las condiciones experimentales no son las mismas que las existentes en la relación terapéutica: el paciente puede mantener una relación de carácter no fiduciario con el investigador o, por el contrario, puede mantener un vínculo fundado en la extrema vulnerabilidad. Por otra parte, el contexto terapéutico en el que se inscribe el tratamiento experimental posee unas formas y lenguajes muy distintos a los empleados en una relación clínica de tipo convencional. Por último, como el trabajo del investigador puede no estar centrado exclusivamente en la salud del voluntario, la dinámica comunicativa y el manejo del paciente se ven alterados paralelamente.
Estos factores son claves para aprender a configurar un modo comunicativo que logre hacer vislumbrar al paciente las particularidades "no terapéuticas" de la participación en un estudio biomédico, lo que nos conduce directamente al segundo factor relevante respecto a los modos comunicativos: las características del participante que modulan la comprensión de la información que se le facilita.
A este respecto, hay que tener en cuenta múltiples factores: el nivel sociocultural del paciente, su grado de conocimientos médicos, su motivación y el grado de complejidad del estudio, así como del vocabulario empleado en la entrevista y la HIP. Entre muchos consejos prácticos posibles, hay que destacar dos en lo que concierne a la información oral: primero, se debe evitar el error, frecuente, de presentar la información de forma rápida y desestructurada, escatimando las oportunidades de meditarla y hacer preguntas; y, segundo, se deben escoger palabras del vocabulario común y construir frases de sintaxis sencilla.
Un medio para determinar si el potencial colaborador podrá entender la hoja de información consiste en hacerla leer a alguien de la misma educación y edad que aquel al que va dirigido el ensayo; también se puede solicitar a los potenciales participantes que expliquen los procedimientos del ensayo con sus propias palabras; asimismo, se debe proporcionar un tiempo mínimo de 15 minutos entre el momento en que se lee la hoja de información y el momento en que el paciente decide dar su consentimiento; finalmente, en la medida de lo posible, es útil facilitar otros soportes complementarios que hagan la información más accesible: CD-ROM, videos, tebeos, etc.
Por último, el método con el cual se pretende informar a los potenciales participantes de una investigación debe ser sometido a la evaluación del Comité de Ética de la Investigación que esté supervisando el estudio. Además, si se busca mejorar los CI, y de cara a futuros ensayos, es una buena idea adquirir la costumbre de preguntar a los antiguos y nuevos voluntarios sobre la calidad del CI trabajado con ellos. Es muy útil preguntar qué puntos fueron o son los de menor claridad y qué información necesitaría un mayor número de ejemplos.
LA VOLUNTARIEDAD
Un tercer factor importante en el proceso de obtención del CI es comprobar que no existe coerción, amenaza, temor o cualquier otro tipo de influencia indebida sobre el participante. La coacción del investigador para forzar el consentimiento atenta de modo característico contra la voluntad del potencial participante en investigación. Esta coacción se produce, por ejemplo, cuando se exagera intencionalmente el peligro de la enfermedad.
El error opuesto a la coacción es el de la influencia indebida. Se produce cuando se ofrece una recompensa excesiva que eclipsa el interés por el avance científico, esencialmente altruista, que debe motivar el consentimiento del voluntario potencial. En este asunto, hay que atender a las especiales circunstancias del participante potencial, pues algunos ofrecimientos habitualmente inocuos pueden constituir una influencia indebida si el sujeto es especialmente vulnerable.
En tercer lugar, es necesario identificar y evitar las presiones injustificadas. Como hemos comentado anteriormente, las más frecuentes son las relacionadas con la posición de influencia y autoridad que ejercen los médicos que tratan al paciente y a la vez le invitan a colaborar en un ensayo clínico. Por supuesto, es difícil establecer el límite entre la persuasión admisible y la influencia indebida. También se ha dicho que es deber del médico discernir cada caso concreto. En este sentido, el artículo 26 de la Declaración de Helsinki estipula que:
Al pedir el consentimiento informado para la participación en la investigación, el médico debe poner especial cuidado cuando el individuo potencial está vinculado con él por una relación de dependencia o si consiente bajo presión. En una situación así, el consentimiento informado debe ser pedido por una persona calificada adecuadamente y que nada tenga que ver con aquella relación.
Debe tenerse especial cuidado en la obtención del consentimiento cuando las circunstancias del sujeto sean desesperadas. En primer lugar, hay que valorar si sus juicios son lo suficientemente ecuánimes y objetivos. En segundo lugar, hay que estimar si el investigador no está descuidando sus obligaciones de tratamiento en aras de conseguir una terapia potencialmente mejor. Y en tercer lugar, hay que sopesar si la invitación misma no ejerce ya, de hecho, coerción, dado el particular estado del paciente.
Por último, el punto quinto del artículo 7 del Real Decreto, en sintonía con otras normativas, defiende el derecho de todo sujeto participante en un estudio clínico, o de su representante legal, a revocar su consentimiento en cualquier momento sin necesidad de justificación y "sin que de ello se derive responsabilidad o perjuicio alguno".
LA CERTIFICACIÓN
Una cuestión formal pero importante, que también señala el Real Decreto, es la necesidad de redactar dos documentos cara a la obtención del consentimiento: por un lado, la HIP, donde debe quedar constancia de los principios de cantidad y calidad de información ofrecida al potencial colaborador —y que este deberá conservar—; y, por otro, el documento de consentimiento propiamente dicho, conformado por las declaraciones y firmas. Tal como señala el punto 2 del artículo séptimo del Real Decreto, ambos deberán ser entregados y explicados mediante una entrevista previa y personal con uno de los miembros del equipo de investigación.
Si el investigador percibe manifiestamente que el sujeto ha recibido y comprendido toda la información sobre su participación en el estudio y acepta libremente colaborar, es el momento de obtener la certificación que recoja tal voluntad. Como se mencionó, el Real Decreto 223/2004 exige, en su artículo 2, que sea realizada por escrito, fechada y firmada. Solo en el caso de que el sujeto tenga un impedimento para escribir, "el consentimiento podrá otorgarse en casos excepcionales de forma oral en presencia de al menos un testigo". La presencia de testigos se justifica también en casos de sujetos de baja o diferente cultura, personas atemorizadas o que requieren un defensor para sentirse más tranquilas. Es importante señalar estas circunstancias en el documento firmado por el testigo quien, además, podrá indicar los extremos pertinentes. En el caso de que el sujeto no sea capaz de dar su consentimiento, el Real Decreto estipula que "la decisión deberá adoptarse por su representante legal en los términos previstos en el artículo 7" (punto 2).
Si el participante es un menor, además del consentimiento del representante legal es necesario obtener, si es posible, el asentimiento del primero, tras haberle proporcionado toda la información adaptada a su nivel de entendimiento. Tal como queda reflejado en la Directiva 2001 de la Unión Europea, "el investigador o, en su caso, el investigador principal tiene que tener en cuenta el deseo explícito de un menor capaz de formarse una opinión y de apreciar estas informaciones, de negarse a participar o de retirarse en todo momento del ensayo clínico" (Directiva 2001, art. 3 bis, c). Sobre este asunto concreto, el punto tercero del artículo 7 del Real Decreto —dirigido a la regulación de la investigación en sujetos con parcial o total alteración de sus competencias de autonomía— exige la obtención del CI en participantes con al menos doce años de edad y siempre notificando al Ministerio Fiscal dichas autorizaciones. En contraste, el CEI de Estados Unidos recomienda que el asentimiento se tenga en cuenta a partir de los siete años.17 Entre otras recomendaciones, aconsejan que con los niños menores de doce años se utilicen explicaciones muy concretas, en contraste con los de doce o más años, en los que es posible manejar ideas de tipo más abstracto.
Estos mismos requerimientos son aplicables a las investigaciones con adultos sin capacidad de otorgar su CI. Aquí, además de obtener la conformidad de su representante legal, hay que contar, en la medida en que la capacidad y autonomía del voluntario sean estimables, con su deseo expreso. Si esto no fuera posible, es necesario incluir en el documento de consentimiento, al menos, la presunta voluntad del participante. Por supuesto, si el paciente recupera suficiente discernimiento en algún momento del ensayo, el investigador tiene del deber de solicitar el consentimiento para continuar. Un artículo que aporta información complementaria a este respecto es el de Helgesson, titulado "Cómo gestionar el consentimiento informado en los estudios longitudinales cuando los participantes tienen una comprensión limitada del estudio" (34).
Una vez firmados los documentos pertinentes, estos han de archivarse junto con el protocolo del ensayo. Todos ellos han de conservarse durante al menos 15 años según la normativa de la UE. Por su parte, el sujeto también debe retener una copia. Por supuesto, estos procedimientos no deben servir para intimidar al sujeto, ni crear miedo o desconfianza. Por el contrario, hay que procurar transmitir al voluntario que esa es, primeramente, la manera de testimoniar el hecho ético involucrado en el estudio, y solo secundariamente, un modo de proteger tanto al participante como al investigador.
Por último, el investigador debe comunicar al voluntario toda nueva información relevante sobre el estudio (nuevos riesgos, cambios en los procedimientos, prolongación del tiempo del estudio, etc.) para ratificar el CI bajo las nuevas condiciones. En definitiva, el proceso de CI dura hasta el final del estudio y más allá pues, como veremos en el próximo apartado, la gestión de los resultados y el almacenamiento y nuevos usos dados a la información y muestras del paciente siguen siendo competencia del colaborador.
RESULTADOS DE LA INVESTIGACIÓN
En la práctica general se considera que la información de los resultados pertenece al equipo investigador. Es este el que decide el modo, momento y destinatarios a quienes se hacen públicos. Para tal fin se utilizan tres canales habituales: reuniones científicas, revistas especializadas y libros de divulgación. Sin embargo, siendo coherentes con el respeto al participante que otorga el CI, hay motivos para defender que dicho derecho se extiende también a la información sobre los resultados que le afecten. Hacerlos partícipes es una manera de mostrar que los sujetos no son considerados como meros medios, sino también como fines de la investigación.
Este tipo de información presenta, sin embargo, varias características singulares que hacen que su transferencia implique otras tantas dificultades. En primer lugar, requiere el empleo de un tiempo extra del investigador en aras de seleccionar y traducir los resultados de los que ha de tener constancia el voluntario. Esto implica un segundo y no menos importante problema, un coste extra en el presupuesto del ensayo.
En tercer lugar, no siempre es sencillo saber cuál es el momento más adecuado, dentro del proceso de la investigación, para ofrecer los resultados. Precipitarse a compartir los datos sin haberlos contrastado y valorado adecuadamente puede dar falsas ilusiones o motivos de preocupación vanos. Por el contrario, esperar hasta el final del proyecto tal vez implique la pérdida de un tiempo precioso a sujetos que quizá hayan puesto en el ensayo sus últimas esperanzas.
En cuarto lugar, hay que tener en cuenta en qué medida los resultados ofrecidos pueden perjudicar, no solo el buen desarrollo del ensayo, sino también al voluntario mismo. El sujeto puede sufrir estrés psicológico durante el ensayo si conoce que, por ejemplo, están apareciendo varios inconvenientes en el proceso de estudio o que los resultados obtenidos hasta la fecha son pobres. Por último, es muy controvertido el beneficio que pueda causar la comunicación de resultados que aporten malas noticias a los sujetos de estudio. Este es el caso, por ejemplo, de ensayos que investigan pruebas de diagnóstico precoz en enfermedades degenerativas hasta la fecha incurables. Por contraste, varias son las ventajas que pueden derivarse de hacer partícipes de los resultados a los sujetos. En primer lugar, el evidente beneficio directo y personalizado que puede conllevar la información sobre cada uno de los individuos que colabora en el ensayo. Véase el caso, por ejemplo, del hallazgo de un nuevo factor de riesgo de desarrollar una enfermedad dentro del grupo de estudio. Dicho dato, acompañado de la subsiguiente recomendación de más frecuentes revisiones periódicas que las aconsejadas a la población general, supondría un beneficio neto. Por el contrario, sería injusto que los sujetos que han hecho posibles tales conclusiones fuesen los últimos en disfrutar de dichos consejos preventivos. En segundo lugar, no hay que presuponer que siempre la información negativa sobre diagnóstico o pronóstico va a perjudicar la situación presente o futura del paciente. Varias investigaciones sobre el test predictivo para la enfermedad de Huntington (EH) apoyan este punto. Estas parecen demostrar que el conocimiento de poseer un alto riesgo de contraer tal enfermedad no se acompaña, como cabría de esperar, de un mayor índice de estrés o depresión, sino todo lo contrario. Este fenómeno puede estar relacionado con la disminución del grado de incertidumbre que acompaña la vida de estos sujetos, así como con una mayor oportunidad de planificación (35, 36).
Por supuesto, el sujeto tiene derecho a declinar conocer la información que crea nociva y el investigador debe respetar su decisión. Este tipo de especificaciones deben aparecer especialmente indicadas en la entrevista y en la hoja de CI de aquellos ensayos clínicos relacionados con pruebas diagnósticas predictivas.
Por otra parte, los resultados de un estudio no tienen por qué ser transmitidos solo a los participantes que se benefician directamente de esa investigación. Como se comenta en un artículo de Fernández, Kodish y Weijer (37), dar a conocer las conclusiones también a quienes no han conseguido superar, por ejemplo, la fase I de un ensayo con quimioterápicos, o a los familiares de voluntarios fallecidos, supone un beneficio indirecto al que también se debiera tener derecho. Además, saber en qué modo se ha colaborado en el progreso científico no solo favorece la autoestima o da valor al sufrimiento presente o pasado, propio o ajeno, sino que además fomenta la confianza en los investigadores, en la investigación y en las instituciones que las sostienen, lo cual revierte en beneficio de la ciencia.
Como síntesis final de la protocolización del CI, cabe citar cuatro consejos prácticos relacionados con este y los resultados de la investigación.
Respecto a la entrevista personal y a la HIP sería conveniente añadir en ellas:
Qué debe hacerse con la información obtenida del estudio.
Qué planes se sugieren, si hay alguno, una vez terminado el ensayo.
Y por lo que se refiere a la comunicación de las conclusiones del estudio:
Hay que tratar de ofrecer los resultados de manera comprensible y personalizada. Es preferible una entrevista cara a cara con uno de los responsables de la investigación.
Hay que subrayar el grado de validez y extensión científica de los resultados.
EXCEPCIONES AL CONSENTIMIENTO INFORMADO
El punto cuarto del artículo séptimo del Real Decreto delimita dos alternativas que liberan al investigador de la obtención del CI, siempre bajo la condición de que tenga un específico interés terapéutico para la población en la que se realiza la investigación. Estas son:
- Riesgo grave sin alternativa terapéutica y sin posibilidad de obtener el asentimiento del paciente o representante legal.
- Que el paciente no pueda tomar decisiones y carezca de presentante legal.
Obviamente, en ambas situaciones se presume que se ha tratado previamente de contactar y obtener el CI de personas vinculadas por razones familiares o de hecho con el paciente.
La última excepción posible, aunque más controvertida, es la relacionada con investigaciones donde se aplica el método de Zelen (38). Este consiste en obtener el CI tras la aleatorización y solo de aquellos que han sido asignados al grupo del tratamiento experimental. Por el contrario, los que reciben el tratamiento convencional no son informados. Sin embargo, el método de Zelen tiene una importante objeción. Choca con el derecho del sujeto a ser informado sobre la aleatorización. Por otra parte, esto puede crear cierta desconfianza en la relación médico-paciente, cuyo fin debería ser siempre y, en primer lugar, la salud de este último.
Además, hay que mencionar ciertos casos que justifican, en el proceso del CI, el ofrecer información incompleta. Tres son los requisitos que tienen que darse simultáneamente para la omisión de datos:
Los objetivos de la investigación se verían modificados al facilitar tal información.
La información revelada no es esencial (riesgos mínimos).
Existe un plan adecuado para retirar a los sujetos cuando sea necesario y para que los resultados de la investigación les sean de provecho.
En todo caso, se debe diferenciar claramente cuál es la información que puede afectar o invalidar el ensayo, del resto de datos que simplemente hacen más fácil la cooperación de los sujetos o que incomodan al investigador. Solo el primer tipo de información tiene justificada su ocultación.
Finalmente, existen otras circunstancias que justifican prescindir de la toma de CI. Un primer caso es la investigación observacional que no supone riesgo físico para el participante. Otro tipo de estudio donde se cumple la excepción es el de las investigaciones sobre comprensión —por ejemplo, de textos de consentimiento informado—.
Una tercera excepción es la de los estudios sobre urgencias extremas o situaciones catastróficas. En este caso, debe existir un plan general de actuación claro aprobado por el Comité de Ética. Es evidente que no todas las urgencias justifican la ausencia de solicitud de consentimiento del sujeto. Para tomar la decisión más acertada es necesario considerar tres factores:
El conflicto existente entre la necesidad del pronto inicio de tratamiento y el tiempo requerido en el proceso de obtención del CI.
La validez del consentimiento del paciente en circunstancias extremas.
Cómo se debe aplicar la renuncia, si procede, a la obtención del consentimiento.
El punto c) está relacionado con el hecho de que, si bien los investigadores pueden desestimar inicialmente la obtención de CI, eso no significa que tal decisión sea válida durante todo el periodo de la investigación. Para más información sobre este tema véase el artículo "Consentimiento informado para tratamiento médico e investigación: una revisión" (39).
REFLEXIÓN CONCLUSIVA
Toda esta normativa sobre el consentimiento informado que hemos examinado detalladamente nos muestra muchos aspectos de la ética de la investigación clínica, tantos, que puede parecer exhaustiva. Sin embargo, ya hemos mencionado, a propósito de las causas del auge de la formalización en tiempos recientes, cómo un protocolo o guía, a la vez que facilita diagnósticos y toma de decisiones, tiene el riesgo de frenar la reflexión personalizada de la situación que nos ocupa. Quien sale perdiendo es el paciente (y también en cierta medida el médico), pues los detalles personales de su situación concreta no tienen por qué aparecer en el protocolo. Sin embargo, vimos al inicio que el diálogo con el paciente para hacerse cargo de sus peculiaridades personales es vital para poder desarrollar a continuación una atención adecuada al caso. La aplicación de las guías o los protocolos, por tanto, lleva a una atención tendente a la rutina que no hace justicia al caso concreto en que nos encontramos.
Esto mismo se podría expresar de modo más genérico: en ciencia, las cuestiones metodológicas son muy peculiares. Por una parte, al sistematizar un método de estudio, permiten elaborar hipótesis que pueden ser comprobadas y hacer avanzar los conocimientos en esa área. Pero, por otra parte, un método de estudio sistematizado, por definición, se centra en la parte de la realidad que se puede abarcar con dicho método. La consecuencia es que las hipótesis que se pueden plantear vienen, de entrada, recortadas por el método, y no se pueden referir a aspectos de la realidad que no han sido contemplados por esa reducción metodológica. Otro tanto sucede con cuestiones de menos nivel: también las hipótesis científicas vigentes modulan las nuevas teorías que se pueden elaborar.18 El resultado es el mismo: solo aparece la parte de realidad que deja visible el método o las hipótesis vigentes. El resto de la realidad se esconde.
Paralelamente, el método formalizado en la investigación clínica deforma la realidad que se quiere observar y bloquea una parte, de modo que la aplicación del protocolo normalizado hace desaparecer lo que no está formalizado en él. Esto no es especial inconveniente mientras el médico sea consciente de que la aplicación del protocolo está recortando la realidad. Por desgracia, muchas veces el investigador piensa que la aplicación correcta de la normativa le permite tener en cuenta toda la situación. La consecuencia es que, al no desvelar parte de la realidad, su atención al paciente no será la adecuada. Tras la formalización, la persona no comparece.19 Y es vital que comparezca, pues sus peculiaridades humanas son las que nos hacen llevar nuestra acción terapéutica por unos u otros derroteros.
En suma, podemos afirmar que la protocolización del consentimiento informado en investigación concreta y precisa bastantes detalles que se deben tener en cuenta en una investigación clínica éticamente correcta, con lo que aporta una base para un comportamiento adecuado. Pero, también, hay que admitir que los protocolos se aceptan ampliamente por motivos en buena medida psicológicos, derivados del sentimiento de seguridad que proporcionan tanto a voluntarios como a investigadores. Y que, a la vez, la protocolización limita seriamente el conocimiento de la realidad vital del paciente, que solo se puede alcanzar por otros medios, como pueden ser una entrevista no estructurada o una narrativa.
3 "El médico [...] estudia la enfermedad desde sus comienzos y según sus fundamentos naturales, cambia impresiones con el mismo enfermo y con los amigos y allegados de este y, al mismo tiempo que él personalmente aprende junto a los enfermos, va instruyendo al mismo paciente, en la medida en que ello es posible, sin prescribirle nada hasta tanto haya conseguido convencerle de ello; y entonces, ayudado ya por la persuasión, tranquiliza y prepara continuamente a su enfermo, hasta lograr llevarlo poco a poco a la salud. ¿Será realmente esta la manera en que el médico aplicará en mejores condiciones el arte de curar [...]? ¿Será procurando este resultado único por medio de los dos procedimientos, o lo hará sirviéndose de uno solo, el peor de los dos, el que molesta más al paciente?" (1).
4 Véase texto de la nota 1, que, en su sencillez, es muy denso en ideas.
5 Es lo que plantea Platón poco antes, comentando cómo practican los esclavos de los médicos con otros esclavos la poca medicina que han aprendido de modo empírico de sus amos: solo las órdenes terapéuticas, sin aportar razones, y sin ciencia.
6 Así, un ilustrado tan serio como Kant, tras elaborar una densa teoría ética y hacer un extraordinario hincapié en el deber puro, escribe un librito, La paz perpetua, en que afirma que, de hecho, los ciudadanos (en el sentido moderno, es decir, solo individuos) se comportan siempre mal, y la única solución para que la sociedad funcione consiste en un conjunto de normas externas coactivas que hagan que la gente se comporte adecuadamente aunque sean demonios (es la expresión original de Kant). No hace falta insistir en lo utópico de esta solución.
7 Esto es falso, como lo demuestran palmariamente los diversos intentos de extraer el altruismo a partir de impulsos meramente egoístas. Un ejemplo se puede encontrar en el planteamiento de Richard Dawkins en El gen egoísta (4), dentro de unas coordenadas evolucionistas radicales. En esta misma línea se mueve la sociobiología de Wilson. En economía, ya es bien conocido el dilema del prisionero. Y, dentro del campo de la bioética, se puede ver la versión liberal radical de H. T. Engelhardt en The Foundations of Bioethics (5), que plantea la sanidad como un mero intercambio comercial para beneficio personal de los contratantes.
8 Los aditamentos posteriores no ilustrados que intentan fundamentar esa defensa del más débil haciendo referencia a la dignidad humana o, con terminología más moderna, a los derechos humanos (entendidos como algo entitativo), son solo eso, aditamentos, que resultan extraños a la idea ilustrada de partida. Solo tienen coherencia en un contexto de ideas no moderno.
9 Marcia Angell ha sido directora del New England Journal of Medicine, y es actualmente catedrática emérita en la Universidad de Harvard.
10 Esta pregunta, que cualquier clínico ha oído miles de veces, no implica que el paciente deje su decisión en manos del médico (cosa que también sucede con frecuencia); cuando el paciente delega en el médico, este pasa, con horror de los modernos, a ser paternalista. Sin embargo, esa pregunta apunta a otras cuestiones, que la visión moderna de relación entre extraños no ve ni por asomo: comprensión del modo de vida del paciente, y un cierto "paternalismo bueno" (7).
11 Es una posible traducción del neologismo, que es más fácil en lengua inglesa (8).
12 Hay una buena traducción castellana online (9). El texto original y otras traducciones son muy fáciles de encontrar en la red.
13 "9. En el curso del experimento el sujeto será libre de hacer terminar el experimento, si considera que ha llegado a un estado físico o mental en que le parece imposible continuar en él. 10. En el curso del experimento el científico responsable debe estar dispuesto a ponerle fin en cualquier momento, si tiene razones para creer, en el ejercicio de su buena fe, de su habilidad comprobada y de su juicio clínico, que la continuación del experimento puede probablemente dar por resultado la lesión, la incapacidad o la muerte del sujeto experimental".
14 Debemos al doctor Gonzalo Herranz estas ideas sobre Nüremberg y algunas sobre la Declaración de Helsinki (10).
15 Existe versión online completa bilingüe (11).
16 Existe versión online completa bilingüe de la última revisión (12).
17 Se puede encontrar más información práctica sobre este tema en la página de los institutos nacionales de salud (NIH) de los Estados Unidos (33).
18 Véase La mente del universo (40), especialmente el apartado "Factores convencionales en la ciencia", pp. 266 y ss.
19 Como muestra de esta incomparecencia, basta mencionar que, en los protocolos examinados, en ningún momento se pregunta al paciente por qué desea participar en la investigación, pregunta que sería la primera en un contexto de trato normal con una persona.
REFERENCIAS
1. Platón. Obras completas. 2a ed. Madrid: Aguilar; 1986.
2. Pardo A. ¿Qué es la salud? Revista de Medicina de la Universidad de Navarra, 1997;41(2):4-9 [visitado 2014 Mar 12] Disponible en: http://www.unav.es/humbiomedicas/apardo/salud.pdf
3. Laín Entralgo P. La curación por la palabra en la antigüedad clásica. 2a ed. Barcelona: Anthropos; 1987.
4. Dawkins R. El gen egoísta. Barcelona: Salvat; 1986.
5. Engelhardt HT Jr. The Foundations of Bioethics. New York: Oxford University Press; 1986.
6. Angell M. Del paternalismo al abandono del paciente (o sobre la perversión del consentimiento informado). [visitado 2009 Oct 17]. Disponible en: http://bioeticayderechosanitabioeticayderechosanitario.blogspot.com/2007/03/del-paternalismo-al-abandono-del.html Es traducción de un artículo de opinión publicado en: Disponible en: http://www.boston.com/news/globe/editorial_opinion/oped/articles/2007/03/05/longing_for_days_when_doctors_still_advised/
7. Pardo A. Más allá de la autonomía. Revista de Medicina de la Universidad de Navarra. 2003;47(3):45-8 [visitado 2014 Mar 31] Disponible en: http://www.unav.es/humbiomedicas/apardo/autonomia.pdf
8. Sokol D. Let's stop consenting patients. BMJ. 2014;348:g2192.
9. Tribunal Internacional de Nüremberg. El "Código" de Nüremberg [visitado 2014 Abr 1]. Disponible en: http://www.unav.es/cdb/intnuremberg.html
10. Rodríguez Sendín JJ, et al. Desde el corazón de la medicina: homenaje a Gonzalo Herranz. Madrid: OMC; 2013.
11. Comisión Nacional para la protección de los sujetos humanos de investigación biomédica y comportamental. Principios y guías éticos para la protección de los sujetos humanos de investigación [visitado 2014 Abr 1]. Disponible en: http://www.unav.es/cdb/usotbelmont.html
12. Asociación Médica Mundial. Declaración de Helsinki de la AMM - Principios éticos para las investigaciones médicas en seres humanos [visitado 2014 Abr 1]. Disponible en: http://www.unav.es/cdb/ammhelsinki2.html
13. Lewis CS. La abolición del hombre. Madrid: Encuentro; 1990.
14. Curran W, Hall M, Kaye D. Health Care Law, Forensic Science, and Public Policy. 4th ed. London: Little Brown and Company; 1990.
15. Lederer SE. Subjected to Science: Human Experimentation in America Before the Second World War. Baltimore: Johns Hopkins University Press; 1995.
16. Bean WB. Landmark perspective: Walter Reed and yellow fever. JAMA 1983;250:659-62.
17. Vollmann J, Winau R. Informed consent in human experimentation before the Nüremberg Code. BMJ 1996;313:1445-49.
18. Trials of War Criminals Before the Nüremberg Military Tribunals Under Control Council. 1949;10(2):181-2.
19. World Medical Association. Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. JAMA. 2013;310(20):2191-94.
20. Beecher HK. Ethics and clinical research. N Eng J Med 1966;274:1354-60.
21. Brandt AM. Racism and research: The case of the Tuskegee Syphilis Study. Hastings Cent Rep 1978;8:21-29.
22. Protection of human subjects. Belmont Report: Notice of report for public comment. Fed Regist. 1979;44:23191-7.
23. Council for International Organizations of Medical Sciences (CIOMS), World Health Organization (WHO). International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva: CIOMS; 2002 [visitado 2015 May 18]. Disponible en: http://www.cioms.chimages/stories/CIOMS/guidelines/guidelines_nov_2002_blurb.htm
24. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for good clinical practice. Geneva, 1996 [visitado 2015 May 19]. Disponible en: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf
25. US Department of Health & Human Services. Protection of human subjects. Subpart A. Basic HHS Policy for Protection of Human Research Subjects (Common Rule). Washington, 2009 [visitado 2014 Abr 1]. Disponible en: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html#subparta
26. Council of Europe. Additional Protocol to the Convention on Human Rights and Biomedicine, concerning Biomedical Research. Strasbourg, 2005 [visitado 2015 May 19]. Disponible en: http://conventions.coe.int/Treaty/en/Treaties/Htmy195.htm
27. Gobierno de España. Real Decreto por el que se regulan los ensayos clínicos de productos farmacéuticos y preparados medicinales. Boletín Oficial del Estado, 108 (6 de mayo de 1978): 10683-4 [visitado 2015 May 19]. Disponible en: http://www.boe.es/buscar/doc.php?id=BOE-A-1978-12157
28. Gobierno de España. Ley 25/1990, de 20 de diciembre, del Medicamento. Boletín Oficial del Estado, 306 (22 de diciembre de 1990): 38228-46 [visitado 2015 May 19]. Disponible en: http://www.boe.es/buscar/doc.php?id=BOE-A-1990-30938
29. Gobierno de España. Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos. Boletín Oficial del Estado, 114 (13 de mayo de 1993): 14346-64 [visitado 2015 May 19]. Disponible en: http://www.boe.es/buscar/doc.php?id=BOE-A-1993-12483
30. Gobierno de España. Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Boletín Oficial del Estado, 33 (7 de febrero de 2004): 5429-43 [visitado 2015 May 19]. Disponible en http://www.boe.es/diario_boe/txt.php?id=BOE-A-2004-2316
31. Gobierno de España. Ley 14/2007, de 3 de julio, de Investigación biomédica. Boletín Oficial del Estado, 159 (4 de julio de 2007): 28826-48 [visitado 2015 May 19]. Disponible en http://www.boe.es/buscar/doc.php?id=BOE-A-2007-12945
32. Western Institutional Review Board. A Guide for Researchers. Puyallup: WIRB; 2015, 169 [visitado 2015 May 19]. Disponible en http://www.wirb.com/Documents/Guide%20for%20Researchers.pdf
33. US Department of Health & Human Services. National Institutes of Health (NIH) [visitado 2015 May 19]. Disponible en: http://www.nih.gov/.
34. Helgesson G, Ludvigsson J y Gustafsson S. How to handle informed consent in longitudinal studies when participants have a limited understanding of the study. J Med Ethics. 2005;31(11):670-673.
35. Huggins M, Bloch M, Wiggins S, Adam S, Suchowersky O, Trew M, et al. Predictive testing for Huntington disease in Canada: adverse effects and unexpected results in those receiving a decreased risk. Am J Med Genet. 1992;42(4):508-15.
36. Hayden MR, Bloch M, Wiggins S. Psychological effects of predictive testing for Huntington's disease. Adv Neurol. 1995;65:201-10.
37. Fernández CV, Kodish E, Weijer C. Informing study participants of research results: an ethical imperative. IRB: Ethics & Human Research. 2003;25(3):12-9.
38. Zelen M. A new design for randomized clinical trials. N Engl J Med. 1979;300(22):1242-5.
39. Carmen MG, Joffe S. Informed consent for medical treatment and research: a review. Oncologist. 2005;10(8):636-41.
40. Artigas M. La mente del universo. Pamplona: Eunsa; 1999.