Artículo
José Alexander Carreño-Dueñas1
1 Instituto Nacional de Cancerología, Colombia. jcarreno@cancer.gov.co
Fecha de recepción: 2015-12-11
Fecha de envío a pares: 2016-01-22
Fecha de aprobación por pares: 2016-04-19
Fecha de aceptación: 2016-07-14
Para citar este artículo / To reference this article / Para citar este artigo
Carreño-Dueñas JA. Consentimiento informado en investigación clínica: un proceso dinámico. pers.bioét. 2016; 20(2): pp. 232-243. DOI: 10.5294/pebi.2016.20.2.8
|
Resumen En investigación clínica, el consentimiento informado es un documento legal y un mecanismo para respetar la dignidad y proteger los derechos y el bienestar de los sujetos participantes; debe incluir información sobre el propósito de la investigación, la justificación, los riesgos y beneficios, que le permitan a un sujeto decidir voluntariamente su participación. Como es deber del investigador velar por la protección de la vida, la salud, la dignidad, la integridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de los sujetos que enrole en un estudio, tiene que establecer un diálogo permanente con estos para valorar los riesgos y la seguridad de la participación; esta condición se convierte en un proceso dinámico que no comienza ni termina únicamente con la firma del documento, y que no solamente debe trascender la legalidad, sino acercarse a la ética y la legitimidad. Palabras clave: consentimiento informado; sujetos de investigación; experimentación humana; ensayos clínicos controlados; investigación biomédica (Fuente: DeCS, Bireme). |
Abstract In clinical research, informed consent is both a legal document and mechanism for respecting the dignity of participating subjects and protecting their rights and wellbeing. It should include information on the purpose of the research, its justification, and the risks and the benefits involved, so as to enable a subject to decide to participate voluntarily. Because it is the researcher’s duty to ensure protection of the life, health, dignity, integrity, right to self-determination, privacy and confidentiality of the subjects who take part in a study, the researcher must establish a permanent dialogue with them to assess the risks and safety inherent in their participation. This condition becomes a dynamic process that neither begins nor ends when informed consent is signed. It goes beyond legality and becomes a question of ethics and legitimacy. Keywords: Informed consent; research subjects; human experimentation; controlled clinical trials; biomedical research (Source: DeCS, Bireme). |
Resumo Em pesquisa clínica, o consentimento informado é um documento legal e um mecanismo para respeitar a dignidade e proteger os direitos e o bem-estar dos sujeitos participantes; deve incluir informação sobre o propósito da pesquisa, a justificativa, os riscos e benefícios, que permitam a um sujeito decidir voluntariamente sua participação. Como é dever do pesquisador velar pela proteção da vida, da saúde, da dignidade, da integridade, do direito à autodeterminação, da intimidade e da confidencialidade dos sujeitos que se envolvam num estudo, tem que estabelecer um diálogo permanente com estes para valorizar os riscos e a segurança da participação; essa condição torna-se um processo dinâmico que não começa nem termina unicamente com a assinatura do documento, e que não somente deve transcender a legalidade, mas, sim, se aproximar à ética e à legitimidade. Palavras-chave: consentimento informado; sujeitos de pesquisa; experimentação humana; ensaios clínicos controlados; pesquisa biomédica (Fonte: DeCS, Bireme). |
Introducción
Universalmente, el consentimiento informado (CI) se fundamenta en el principio de autonomía y en la libertad de una persona mentalmente competente para aceptar o rechazar cualquier forma de participación, intervención o procedimiento de investigación (1), y se constituye en un acuerdo de voluntades entre el investigador y el sujeto participante, que deberá regirse bajo las dimensiones de confianza, sinceridad, claridad, respeto, ausencia de manipulación, engaño o coerción (2).
El National Center for Biotechnology Information (NCBI) define al CI como la autorización voluntaria de un sujeto de investigación, con una comprensión completa de los riesgos involucrados en la práctica de procedimientos de investigación (3). En el manual de ética médica de la American Medical Association (AMA), el CI es considerado como un concepto central de la ética médica actual y se fundamenta en el derecho de un sujeto a recibir, por parte del médico, la información necesaria para tomar sus decisiones (4).
La investigación clínica busca generar nuevo conocimiento a través de la aplicación de las ciencias naturales, especialmente la biología y la fisiología, a la medicina, para tener una mejor comprensión de la génesis de las enfermedades que permitan mejorar las opciones de prevención, diagnóstico y tratamiento (5); sin embargo, estos intereses nunca deben vulnerar los derechos y la autonomía de los sujetos participantes (6, 7), como quedó establecido en el Código de Nüremberg desde 1946, donde se promulgó por primera vez el consentimiento voluntario de los sujetos participantes como un mecanismo para proteger sus derechos; desde entonces, el consentimiento se constituye en un requisito indispensable para proteger la autonomía de las personas y su decisión voluntaria para participar en investigaciones experimentales con seres humanos (8, 9).
Adicionalmente, con la primera declaración de Helsinki en 1964, se estableció que es deber del investigador velar por la protección de la vida, la salud, la dignidad, la integridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de los sujetos (10). En 1979, con el informe de Belmont, se establecieron las guías éticas para la protección de los seres humanos que participen en investigación experimental, y se definieron los principios fundamentales que debe contener el CI, como son: la información, la comprensión y la voluntariedad (11). En 1995, la Organización Mundial de la Salud (OMS) publicó las guías de buena práctica clínica (GPC) para regular la investigación farmacológica con seres humanos; en estas guías se estableció que los principios del consentimiento informado deben ser implementados en cada investigación (12); de la misma manera, en las guías del Council for International Organizations of Medical Sciences (CIOMS) se estableció como un requisito fundamental para el desarrollo de investigaciones clínicas, que el otorgamiento del CI de un sujeto participante debe ser previo a su participación (13).
En Colombia, con la Resolución 8430 de 1993, se definió el riesgo de las investigaciones clínicas y aquellas que sean consideradas con un riesgo superior al mínimo deberán contar con un consentimiento informado firmado por escrito por el sujeto o por su representante legal. Por tal razón, el CI se convirtió en un documento legal y en un mecanismo para respetar la dignidad, y proteger los derechos y el bienestar de los sujetos participantes de investigaciones (14). En la misma resolución se estableció que el CI debe incluir información sobre el propósito de la investigación, la justificación, los procedimientos que le realizarán al sujeto participante, los riesgos y beneficios, aspectos logísticos y administrativos, entre otros, para que el sujeto, con pleno conocimiento de la naturaleza del estudio a que se someterá, autorice voluntariamente su participación (15). La validez y la calidad de la información que contenga este documento estará sujeta a revisión por parte de los comités de ética de las instituciones que conducen estudios de investigación y del Instituto Nacional para la Vigilancia de Medicamentos y Alimentos en Colombia (Invima) (16); incluso de agencias regulatorias internacionales como la Food and Drug Administration (FDA) (17), la European Union Agency (EMA) (18), la International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) (19).
Con este trabajo buscamos consolidar la información disponible relacionada a fin de que sea de utilidad para los investigadores en mejorar el proceso del consentimiento informado y para los sujetos participantes en la adecuada comprensión de los alcances del mismo. Se revisaron documentos normativos, guías y recomendaciones nacionales e internacionales. Adicionalmente, se hizo una revisión de artículos publicados en inglés y en español a partir de 1990, que contuvieran las siguientes palabras clave: consentimiento informado, sujetos de investigación, experimentación humana, ensayos clínicos controlados, investigación biomédica; se utilizaron operadores booleanos, AND, OR, NOT y sus combinaciones. Las principales bases de datos consultadas fueron: MedLine, EBSCO, The Science Direct, The Cochrane Library, Embase, Lilacs. Los artículos fueron seleccionados con base en el título, el resumen, la fecha de publicación, el idioma y su relación con el tema de revisión.
Desarrollo del tema
Proceso de obtención
Todos los participantes en estudios de investigación pueden retirar su consentimiento informado en cualquier momento sin que esto signifique algún tipo de sanción (10). El otorgamiento del consentimiento no comienza ni termina únicamente con la firma del documento por parte del sujeto, el investigador y los testigos (20). La firma es tan solo el producto final de un proceso que debe ser dinámico y de diálogo permanente entre el investigador y el sujeto participante; este documento no tiene fecha de vencimiento y requiere renovarse cada vez que se realicen cambios al protocolo de investigación o cuando se vea comprometida la seguridad del participante, ya sea porque el sujeto experimenta efectos adversos con la terapia de estudio o porque su salud se pone en riesgo (21).
En la evaluación de seguridad, el investigador debe analizar periódicamente la información de seguridad del estudio que le provee el patrocinador a través de los Serious Adverse Event (SAE) y los Suspected Unexpected Serious Adverse Reaction (SUSAR), y determinar en el sujeto el perfil de seguridad (22). Todas las modificaciones realizadas al protocolo de investigación y la información de seguridad deben ser informadas al sujeto para que de esta manera voluntariamente decida continuar en la investigación y otorgue un nuevo consentimiento, actividad que se conoce comúnmente como reconsentir. Igualmente, el sujeto tiene el derecho de retirar su consentimiento en cualquier momento, sin que esto llegue a afectar la atención médica que venga recibiendo (23).
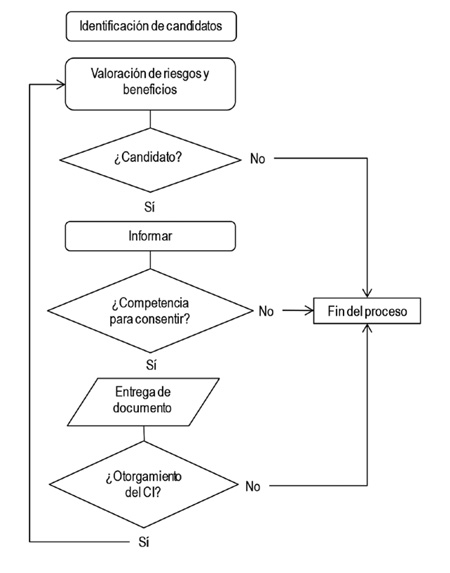
Como se ilustra en la figura 1, el proceso del consentimiento informado empieza cuando un investigador, tras haber identificado un posible participante, le realiza una evaluación de los riesgos y beneficios que represente para su estado salud el participar en un estudio de investigación, para que de esta manera pueda determinar si es viable o no. Si el sujeto es candidato, el investigador deberá informarle sobre los aspectos generales y específicos del estudio; posteriormente, el investigador deberá evaluar la competencia que tiene para consentir y de esta manera hacer entrega del documento y, en caso de que el candidato decida otorgar voluntariamente su consentimiento, proseguir con las respectivas firmas.
Figura 1. Proceso de obtención del consentimiento informado
El proceso de obtención del consentimiento informado merece toda la atención, rigurosidad y respeto —como si se tratase de una consulta médica—, por lo que debe hacerse en un ambiente físico adecuado y privado. El investigador debe permitir un diálogo y destinar el tiempo necesario para explicar al sujeto en qué consiste la investigación. En caso de que el candidato tenga interés en participar, se le deberá suministrar una copia escrita del consentimiento en el idioma que este hable, y permitirle a él o a su representante legal el tiempo suficiente para que lo consulte con sus familiares o allegados. En caso de que el sujeto decida participar voluntariamente en el estudio, deberá firmar el documento en presencia del investigador y de dos testigos que el participante considere pertinentes, o los que la institución provea.
Principios fundamentales del consentimiento informado
Estos principios fueron establecidos de tal manera que estuvieran contenidos en el documento escrito; sin embargo, estos mismos elementos deben ser utilizados por el investigador para la obtención inicial del CI y durante el desarrollo de la investigación.
La información
Uno de los elementos fundamentales que favorece el entendimiento de los sujetos del consentimiento informado es la información que le provea el investigador (24); esta debe contener los elementos de calidad dispuestos por la declaración de Helsinki (25), y es considerada como un derecho que favorece la toma de decisiones de los sujetos sobre participar en un estudio de investigación (26).
El investigador debe apoyarse en los principios éticos y científicos que rigen en toda nueva investigación médica; debe revisar la investigación preclínica y la evidencia científica disponible que justifique su realización con seres humanos (27). Toda la información científica, la justificación, los objetivos y el diseño metodológico del estudio deberá ser completamente conocida y comprendida por el investigador de tal manera que pueda responder a las preguntas que surjan tanto por los comités de ética de las instituciones donde se conducirá el estudio como por los sujetos participantes (22).
La información que proporcione el investigador al sujeto deberá tener un lenguaje sencillo, sin tecnicismos y de fácil comprensión (28), pero con claridad y prudencia frente a las posibilidades de respuesta, curación, recaída o pronóstico de supervivencia; así mismo, deberá ser suficiente y precisa respecto a los riesgos y beneficios del estudio (29).
Aspectos generales
Deberá hacer una descripción general del estudio que incluya:
En qué consiste el estudio de investigación.
Cuáles son los propósitos y la justificación de realizarlo.
Descripción de la terapia de investigación y explicación de los mecanismos de acción.
Duración estimada de la participación del sujeto y la duración total del estudio.
Descripción detallada de la frecuencia de exámenes y procedimientos que se le realizarán.
Explicar cómo la institución y el equipo de investigación mantendrán la confidencialidad de sus datos personales y de su información clínica.
Explicar cómo se protegerá al participante en caso de que ocurran lesiones o daños resultantes de la participación y la previsión de atención médica inmediata.
Informar al sujeto si va a recibir algún tipo de compensación económica por participar (30).
Explicar cómo se indemnizará al paciente en caso de sufrir una discapacidad secundaria a la terapia del estudio, y la existencia de pólizas y compensaciones legales con que cuenta la institución donde se realizará la investigación.
Explicar las razones por las cuales se podría dar por terminada la participación en el estudio.
Riesgos y beneficios
Se debe realizar una clara y completa descripción de los riesgos y beneficios de participar en el estudio, que incluya:
Frecuencia de ocurrencia de riesgos físicos y psicológicos.
Riesgos sociales debidos a posibles rupturas en la confidencialidad que lleven al estigma o a la discriminación social.
Riesgos económicos, tales como gastos de viajes o tiempo sin trabajar.
Riesgos mínimos y habituales, tales como incomodidad al tomar muestras de sangre.
Riesgos severos, discapacidades e incluso la muerte.
Beneficios directos que incluyan aquellos directamente relacionados con los resultados del estudio. Por ejemplo, beneficios por los efectos de un estudio de diagnóstico o un agente experimental.
Beneficios indirectos vinculados a la salud como obtener mejores cuidados o atención extra por parte del equipo de investigación.
Beneficios generales como cuando sus acciones producirán una mejora a otras personas.
Otros beneficios como los que se presentan a la sociedad debido a las perspectivas de tener una población más saludable.
Terapias y procedimientos del estudio
De acuerdo con el diseño del estudio, debe explicar las terapias incluidas en cada brazo de tratamiento de la investigación, cómo se garantizará una distribución equitativa y la posibilidad de asignación aleatoria a uno de los brazos (31). Informar sobre alternativas disponibles de tratamiento distintas a las del estudio de investigación que puedan resultar beneficiosas para el sujeto y, cuando aplique, explicar qué es un placebo y por qué se incluye en el estudio (32, 33).
Comprensión
La comprensión le permite al investigador determinar la competencia que tiene un sujeto para consentir (11). El proceso de otorgamiento del consentimiento informado requiere de tiempo, por lo que el investigador debe permitirle al sujeto que consulte con sus familiares para que tome una decisión y, en caso de que manifieste dudas, responderlas adecuadamente (34).
Se debe tener presente que algunos factores como el nivel educativo y el estrato socioeconómico afectan de una manera directa la forma como un paciente comprende y entiende las indicaciones impartidas por un equipo de salud (35, 36), de tal manera que el investigador debe determinar la capacidad de decisión del sujeto identificando:
Habilidad de comprensión y capacidad de decisión.
Identificar algún desorden mental, lesión cerebral adquirida o demencia.
Identificar la presencia de discapacidades intelectuales.
Debe explicar los deberes y las responsabilidades del sujeto en la investigación
El investigador debe evaluar si el sujeto ha comprendido y captado adecuadamente la información sobre los aspectos de la investigación, y puede utilizar una prueba oral o escrita; esto le permitirá determinar el grado de comprensión del participante y si está en capacidad de ejercer plena autonomía en su decisión. En caso de que el sujeto tenga una pobre comprensión de la información, el investigador debe enfocar sus esfuerzos en mejorar los aspectos que no fueron entendidos o que son confusos (20, 24).
Vulnerabilidad
Implica el deber de brindar una protección adicional a poblaciones especiales que no tengan la capacidad de proteger sus propios intereses o cuando se encuentran bajo circunstancias específicas que las convierten en vulnerables (37). Algunos grupos que pueden ser considerados vulnerables, pero que no necesariamente lo son, pueden ser:
Grupos y comunidades étnicas, especialmente cuando su lenguaje sea distinto al del investigador, en este aspecto deberá revisar cuidadosamente la traducción de los documentos y su significado (38).
Población infantil, a quienes la información les deberá ser suministrada en un lenguaje sencillo y fácil de entender (39).
Grupos de edad muy avanzada (40).
Personas con enfermedades críticas o incurables y en estados terminales (41).
Poblaciones que estén de alguna manera subordinadas, como fuerzas militares.
Voluntariedad
Se constituye en el elemento que valida la participación de un sujeto en una investigación y debe reflejar condiciones libres de influencias y coerciones indebidas (42, 43). El respeto por la autonomía se fundamenta en la confianza y la cooperación que se establezca entre el investigador y el sujeto participante (44).
El investigador debe explicar la voluntariedad de la participación, debe responder a las expectativas que le surjan (45, 46) y aclarar que puede retirar su consentimiento en cualquier momento sin que exista alguna penalidad o pérdida de beneficios a los cuales el sujeto tiene derecho.
Se deben evitar factores que interfieran con la voluntad del paciente (47) como:
Realizar amenazas de daño a la salud si no participa.
Establecer inminencia de muerte si no acepta el tratamiento.
Realizar incentivos inadecuados o recompensas excesivas (ofrecer dinero, objetos, etc.).
Realizar falsas promesas o expectativas frente a los resultados del estudio.
Se deben evitar actitudes que provoquen coerción en la decisión del sujeto (48) como:
Presiones injustificadas.
Ejercer algún tipo de autoridad.
Realizar algún tipo de manipulación emocional.
Conclusiones
En investigación clínica se debe hacer una clara distinción entre legalidad y legitimidad, porque no todo lo legal es legítimo. El cumplimiento de los requisitos dentro de un marco regulatorio y la obtención del consentimiento informado favorecen la legalidad en investigación clínica y son una condición que está vigilada por las agencias regulatorias. Pero la legitimidad, vista como la conformidad con los principios éticos en la investigación, es una responsabilidad que enfrentan los comités de ética y el investigador, quien es el garante de la seguridad al ejercer un rol de protector de la salud de los sujetos participantes (49), y aunque el CI sea considerado como un requisito ético, no todas las veces es suficiente para que una investigación clínica sea ética (50).
El investigador clínico tiene el deber de velar por la protección de la vida, la salud, la dignidad, la integridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de los sujetos que enrole en un estudio; en tal virtud, la dimensión que adquiere el consentimiento informado no termina con la firma del documento por las partes involucradas. El otorgamiento del consentimiento informado que hace un sujeto es un voto de confianza hacia el investigador y, en contraprestación, este debe mantener un proceso continuo de valoración de riesgos para la seguridad del sujeto participante a medida que la investigación avanza y así determinar la permanencia del sujeto en el estudio (51). Esto convierte al CI en un proceso dinámico de permanente diálogo y evaluación de riesgos.
REFERENCIAS
1. Dhar H, Dhar D. Informed consent in clinical practice and literature overview. Arch Gynecol Obstet. 2012;286(3):649-651.
2. Saavedra J. Consentimiento informado. Rev Médico Leg. 1999;V(1):9-20.
3. The National Center for Biotechnology Information (NCBI). Informed Consent; 2015 [visitado 2015 jun 15]. Disponible en http://www.ncbi.nlm.nih.gov/mesh/68007258
4. World Medical Association. Manual de ética médica, 3a edición. Ferney-Voltaire Cedex, Asociación Médica Mundial; 2015.
5. Houghton Mifflin Harcourt. The American Heritage Dictionary. Boston: Houghton Mifflin; 2015.
6. Claessens MT, Bernat JL, Baron JA. Ethical issues in clinical trials. Br J Urol. 1995;76(Suppl 2):29-36.
7. Tuthill KA. Human experimentation. Protecting patient autonomy through informed consent. J Leg Med. 1997;18(2):221-250.
8. Annas GJ, Grodin MA. The nazi doctors and the Nuremberg Code Human Rights in Human Experimentation. New York: Oxford University Press; 1992 Monograph Collection (Matt - Pseudo); 1992.
9. Vollmann J, Winau R. Informed consent in human experimentation before the Nuremberg Code. Br Med J. 1996;313(7070):1445-1449.
10. World Medical Association. Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects; 2013.
11. National Commission for the Protection of Human of Biomedical and Behavioral Research. The Belmont Report. 1979.
12. World Health Organization. Guidelines for good clinical practice (GCP) for trials on pharmaceutical products [Internet]; 1995 [visitado 2015 jun 15]. Disponible en http://apps.who.int/prequal/info_general/documents/GCP/gcp1.pdf
13. International ethical guidelines for biomedical research involving human subjects. Bull Med Ethics. 2002;(182):17-23.
14. República de Colombia Ministerio de Salud. Resolución 8430 de 1993.
15. Corte constitucional de Colombia. Sentencia T-401/94, Derechos del paciente-autonomía/derecho a la salud-conflictos médico paciente. Bogotá: Corte Constitucional; 1994.
16. Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima) [Internet]. Disponible en https://www.invima.gov.co/
17. U.S. Food and Drug Administration [visitado 2015 ene 1]. Disponible en http://www.fda.gov/
18. Science Medicines Health. European Medicines Agency [Internet]. Disponible en http://www.ema.europa.eu/ema/
19. The International Conference on Harmonisation [Internet]. Disponible en http://www.ich.org/
20. Flory J, Emanuel E. Interventions to improve research participants’ understanding in informed consent for research: a systematic review. JAMA. 2004;292(13):1593-1601.
21. Helgesson G, Eriksson S. Does informed consent have an expiry date? A critical reappraisal of informed consent as a process. Camb Q Healthc Ethics. 2011;20(1):85-92.
22. International Conference on Harmonitation (ICH). Guideline for good clinical practice. United States of America: ICH; 1996.
23. National Institute of Mental Health (NIH). Guía para los participantes sobre investigaciones clínicas de la salud mental; 2015 [visitado 2015 sep 24]. Disponible en ahttp://www.nimh.nih.gov/health/publications/espanol/gu-a-para-los-participantes-sobre-investigaciones-cl-nicas-de-la-salud-mental/index.shtml#pub2
24. Nishimura A, Carey J, Erwin PJ, Tilburt JC, Murad MH, McCormick JB. Improving understanding in the research informed consent process: a systematic review of 54 interventions tested in randomized control trials. BMC Med Ethics. 2013;14:28.
25. Lynoe N, Sandlund M, Dahlqvist G, Jacobsson L. Informed consent: study of quality of information given to participants in a clinical trial. BMJ. 1991;303(6803):610-613.
26. Brigard D, María A. Consentimiento informado del paciente. Rev Colomb Gastroenterol. 2004;19(4):277-280.
27. Daicos GK. Ethical dilemmas encountered during clinical drug trials. Hum Health Care. 2001;1(2):E9.
28. Christenbery TL, Miller MR. A strategy for learning principles and elements of informed consent. Nurse Educ. 2008;33(2):75-78.
29. Agre P, Campbell FA, Goldman BD, Boccia ML, Kass N, McCullough LB, et al. Informed Consent: the medium is not the message. Irb a Rev Hum Subj Res. 2003;Suppl:S11-S19.
30. U.S. Food and Drug Administration. Regulatory Information. Payment to research subjects; 2015 [visitado 2015 sep 24]. Disponible en http://www.fda.gov/RegulatoryInformation/Guidances/ucm126429.htm
31. Freedman B. Equipoise and the ethics of clinical research. N Engl J Med. 1987;317(3):141-145.
32. Hrobjartsson A, Gotzsche PC. Is the placebo powerless? An analysis of clinical trials comparing placebo with no treatment. N Engl J Med. 2001;344(21):1594-1602.
33. De La Fuente-Fernandez R, Stoessl AJ. The biochemical bases for reward. Implications for the placebo effect. Eval Health Prof. 2002;25(4):387-398.
34. Montalvo W, Larson E. Participant comprehension of research for which they volunteer: a systematic review. J Nurs Scholarsh. 2014;46(6):423-431.
35. Surbone A. Cultural aspects of communication in cancer care. Support care cancer off. J Multinatl Assoc Support Care Cancer. 2008;16(3):235-240.
36. Zavala-Sarrio S, Gutiérrez W, Chiang M. Seguimiento del proceso de obtención del consentimiento informado en los participantes de protocolos de investigación. Rev Soc Peru Med Interna; 2007 [visitado 2015 jun 15]; 20(1):10-15. Disponible en http://sisbib.unmsm.edu.pe/BVrevistas/spmi/v20n1/pdf/a03v20n1.pdf
37. Luna F. Vulnerabilidad: la metáfora de las capas. Secretaría Distrital de Salud. Capacitaciones Comité de Ética para la Investigación en Salud; 2014 [visitado 2016 jul 05]. Disponible en http://www.saludcapital.gov.co/Paginas2/Capacitaciones_Comitedeetica.aspx
38. Dickert N, Sugarman J. Ethical goals of community consultation in research. Am J Public Health. 2005;95(7):1123-1127.
39. O’Lonergan TA, Milgrom H. Ethical considerations in research involving children. Curr Allergy Asthma Rep. 2005;5(6):451-458.
40. Barron JS, Duffey PL, Byrd LJ, Campbell R, Ferrucci L. Informed consent for research participation in frail older persons. Aging Clin Exp Res. 2004;16(1):79-85.
41. Verastegui EL. Consenting of the vulnerable: the informed consent procedure in advanced cancer patients in Mexico. BMC Med Ethics. 2006;7:E13.
42. Emanuel EJ. Ending concerns about undue inducement. J Law Med Ethics. 2004;32(1):100-105.
43. Sears JM. Context is key for voluntary and informed consent. Am J Bioeth. 2005;5(1):47-58.
44. Visbal-Illera GC. Capacidad para tomar decisiones frente al Consentimiento Informado de personas que potencialmente participarían en estudios clínicos experimentales para la industria farmacéutica. Salud Uninorte. 2010;26(1):1-11.
45. Wray RJ, Stryker JE, Winer E, Demetri G, Emmons KM. Do cancer patients fully understand clinical trial participation? A pilot study to assess informed consent and patient expectations. J Cancer Educ. 2007;22(1):21-24.
46. Kass NE, Maman S, Atkinson J. Motivations, understanding, and voluntariness in international randomized trials. IRB. 2005;27(6):1-8.
47. Marcovitch H. Misconduct by researchers and authors. Gac Sanit. 2007;21(6):492-499.
48. Hawkins JS, Emanuel EJ. Clarifying confusions about coercion. Hastings Cent Rep. 2005;35(5):16-19.
49. Outomuro D. Reflexiones sobre el estado actual de la ética en investigación en Argentina. Acta Bioeth. 2004;10(1).
50. Emanuel E. ¿Qué hace que la investigación clínica sea ética? Siete requisitos éticos. En Lolas-Stepke F, Quezada-Sepúlveda Á. Pautas Éticas de Investigación en Sujetos Humanos: Nuevas Perspectivas. Santiago de Chile: Ed. Programa Regional de Bioética OPS/OMS; 2003.
51. Fried E. Physician duties in the conduct of human subject research. Account Res. 2001;8(4):349-75.